
Comprehensive targeted super-deep next generation sequencing enhances differential diagnosis of solitary pulmonary nodules
Introduction
Lung cancer continues to be the leading cause of cancer mortality in both men and women worldwide (1). Early diagnosis is crucial for improving lung cancer survival, given that the prognosis of stage I lung cancer is considerably favourable with a 5-year survival rate of more than 70% compared with metastatic late-stage disease (<5% survival) (2). Currently, the most successful method for early detection is low-dose computed tomography (LDCT) scan screening, which was demonstrated by the National Lung Cancer Screening Trial (NLST) study to reduce mortality by 20% compared with chest radiograph screening of lung cancer (3).
The widespread application of LDCT has led to a significant increase in the detection of lung nodules (4,5). The prevalence of solitary pulmonary nodules (SPN) (<3 cm in diameter) is 10–20% in the United States (6) and is higher in people with Asian ancestry probably due to genetic and environmental factors. Most SPN found in CT scans are benign, even among high-risk populations such as smokers. A few algorithms or prediction models based on nodule features in the CT scan have been developed; however, their accuracy remains unsatisfactory (7). On one hand, timely identification of malignant nodules is crucial because they represent a localized disease and are potentially curable. On the other hand, it is costly and possibly harmful to manage an SPN with radiation exposure from repeated CT scans or invasive procedures such as biopsy or surgical resection that are associated with potential morbidity and induce unnecessary anxiety. Therefore, there is a critical need for additional tests that can further stratify the SPN found by LDCT as malignant and non-malignant.
Non-invasive tests are preferable. 18F-FDG-PET/CT only slightly adds to diagnostic value, and its use is limited by its low cost-effectiveness (8). A few plasma biomarkers, such as CEA and CA-125, have been used to screen and diagnose lung cancers (9-11). However, the sensitivity of serum biomarkers is relatively low because they are proteins and thereby will be elevated only when the tumour burden is high. Therefore, there is no sufficiently reliable biomarker that exhibits both high sensitivity and specificity for the diagnosis of malignant SPN. ctDNA represents a promising option: it is released or excreted by tumour cells, circulates in the blood of a patient with cancer, and can serve as direct evidence of malignancy (12).
Because of the diverse mutation pattern of lung cancer, it cannot be evaluated using conservative single-gene mutations or hotspot mutations. Unlike PCR-based techniques, NGS simultaneously allows the detection of a wide spectrum of loci. Comprehensive analyses could theoretically increase sensitivity. In addition, genetic mutations should be more reliable than other qualitative markers (e.g., antibody or micro-RNA level), which require tricky cut-offs.
Previously, a report described using the total plasma cell-free DNA (cfDNA) level to discriminate non-small cell lung cancer (NSCLC) from benign lung pathologies and healthy controls with 86.4% sensitivity and 61.4% specificity (13). However, debates remain regarding the lower limit of detection of ctDNA NGS. We hypothesized that the analysis of the lung cancer-related somatic mutations from ctDNA could provide better opportunities for minimally invasive SPN diagnosis. We hereby aimed to develop a practical tool based on ctDNA profiling and super-deep sequencing methods and test its ability to distinguish between malignant and non-malignant SPN in this pilot study. However, debates remain regarding the lower limit of detection of ctDNA NGS.
Results
Lung peripheral nodule clinical features and tumour serum protein marker classification
A total of 1,254 consecutive candidate patients were reviewed for resection of lung peripheral nodules in the First Affiliated Hospital of Guangzhou Medical University. In postoperative pathological examination, 69% of lung peripheral nodules were diagnosed as malignant, and the distribution of malignant SPN subtypes is shown in Figure S1. Almost 80% of SPNs were adenocarcinoma, which included 13% AAH (atypical adenomatous hyperplasia)/AIS (adenocarcinoma in situ)/MIA (minimally invasive adenocarcinoma) malignancy patients. Surgery and biopsy were risky for the patients and could cause complications. A non-invasive method was required to identify the malignancy of surgery-candidate lung peripheral nodules. Tumour serum protein markers (CEA, NSE, CA125, CA153, and CYFRA21-1) are conventionally used to determine the malignancy of SPN. However, only CEA and CYFRA21-1 in malignant SPN were significantly higher than in non-malignant SPN. The expression of NSE and CA153 in this statistical cohort was not different between malignant and benign cases based on the p-value calculated by the unpaired t-test (Figure 1). The expression of CA125 in benign SPN was significantly higher than in malignant SPN. The mean expression of serum protein markers was similar. Therefore, this signature limits serological indicators as an accurate diagnosis of early lung cancer. Neither CEA (cut-off 5 ng/mL, specificity 90.1%, sensitivity 23.8%) or CYFRA21-1 (cut-off 3.3 ng/mL, specificity 80.6%, sensitivity 28.5%) nor their combination (specificity 77.6%, sensitivity 42.1%) could precisely predict malignancy. When using 10 ng/mL as the cut-off, CEA achieved a specificity of 97.1%, but the sensitivity was only 9.7%.


ctDNA seemed to be a good option as it has been largely reported and named as a non-invasive method for patients’ targeted genes tests and recurrent monitoring (14,15). In this study, both surgically resected lung peripheral nodules and plasma DNA were investigated by extra-deep high throughput sequencing of at least 10,000-fold depth to classify malignancies or non-malignancies in the early stage of lung cancer. The pipeline of this research is shown Figure S2. Thirty-five prospective samples were consecutively collected to perform the next generation sequencing (NGS) to develop a non-invasive malignant peripheral nodule prediction method. All the lung surgery candidate nodules, formalin-fixed, paraffin embedded (FFPE) tissues, and corresponding blood samples were collected as controls. Clinical summary information of the selected patients with lung pulmonary nodules is shown in Table 1; 62.9% of the patients were males, and 37.1% were females. Clinical histological results identified malignant peripheral nodules in 31 out of 35 patients and benign peripheral nodules in the remaining 4 patients. Out of 31 malignant SPNs, 81% of patients were diagnosed with lung adenocarcinoma, which had a much higher distribution than our statistical cohort, likely because of the relatively small sample size. All included cases were in clinical stage I. In postoperative pathological evaluation, 83.9% of the patients remained in stage I, while 29% of patients had advanced to stage II and III which were diagnosed postoperatively by incidental finding of positive lymph nodes. Therefore, all cases are necessary to be screened by NGS to explore the genomic profile. Each sample’s detailed clinical information is recorded in Supplementary Table S1.


Full table

Full table
Landscape of somatic mutations and driver genes
DNA from white blood cells was used as a corresponding normal control to detect somatic mutations from FFPE and ctDNA samples. All of the samples were analysed by lung cancer target capture and sequenced by the Illumina HiSeq 2000 instrument. The lung cancer panel included the exon region of lung cancer driver genes and top mutational lung adenocarcinoma-related genes, based on the COSMIC database (Table S2). Followed by deep sequencing, at least 99.9% of the target genomic regions of each case were covered (Table S3). The median depths were 600× (from 171 to 1,941) for the 38 FFPE samples, 823× (from 524× to 2,543×) for the 38 normal control samples, and 1,896× (from 610 to 7,653) for the 35 ctDNA samples.


Full table
In total, 89 non-silent SNV/InDels/SV (range from 0 to 11) were discovered in the 31 tumour tissue samples. No mutations were detected from the 4 benign pulmonary nodules (Figure 2). Twenty-nine of the 31 cancer samples contained at least one non-silent mutation, and non-silent mutations were detected in all of the lung adenocarcinomas. The two samples in which mutations were not detected were lung carcinoid tumours. This might be because of the limitation of the panel’s gene list, which was based on lung adenocarcinoma and was not available for lung carcinoid tumours and squamous carcinoma. Each sample’s detailed mutational information, excluding that of SV, is shown in Table S4. Four fusions were detected in 3 samples, and 2 of them (ALK, ROS1) were found in sample S23_A. ALK was found in sample S5_A, and RET was found in sample S27_A. However, only 3 out of 4 were successfully validated by immunohistochemistry, except ROS1 fusion in sample S23 (Table S5). Twenty-eight non-silent mutations (SNV/InDel) were detected in the corresponding plasma samples. Only 1 non-silent mutation was detected in the plasma of benign sample S32_N (Figure 2, Table S6). Clinical information of each patient is shown in Figure 2, with the distribution of stage, subtypes, sample types, and gender. The malignant SPN were divided into two groups according to the tumour stage in Figure 2 (stage I vs. stage II-III). For the patients with SPN, each patient’s mutational number had no difference in the tissue or plasma samples (Figures 2,S3). Compared with the mutations in the tissue of benign cases, mutations detected in the tumour tissue had a significantly higher mutational ratio. The mutational number from ctDNA was also assessed with respect to the size of SPN or the mutations in tissue, but no correlation was found (Figures 2,S4). After comparison of the mutational consistencies between tissue and plasma (Figure S5), only 6 out of 152 mutations detected in FFPE were found in the corresponding ctDNA samples; the concordance between ctDNA and FFPE samples was much lesser than that in a previously reported study (16). Even more, 5 out of the 6 overlapping mutations came from one sample (S8_A), which shows a lack of efficacy in early stage ctDNA evaluation to some degree.


Full table

Full table

Full table



Well-known driver genes were detected in 22 out of 31 (71%) malignant FFPE samples, and the frequency of each driver gene was as follows: EGFR: 46%, KRAS: 3%, ALK/ROS1/RET fusion: 11%, BRAF: 3%. The frequency of each driver gene was different from that in our previous study (17,18), especially KRAS, which might be because of sample size limitation and the sequencing panel, which was designed only for lung adenocarcinoma. Except for sample S23_A with non-validation ROS1 fusion, all the other SPNs had a unique driver gene (Figure 3). Of the EGFR-positive SPN samples, 37.5% had compound EGFR mutations; 8 samples contained L858R mutations, and 7 samples had exon 19 deletions (Table 2). Even 3 EGFR mutations were found in sample S22_A. Although all the EGFR compound mutations were rare, SNV/InDel and the co-EGFR mutational ratio were higher than in a recent Asian study (19), This previous study proved that patients with a single EGFR mutation had better survival rates than patients with compound EGFR mutations, and there were no differences in disease-free survival rates. All the EGFR co-mutation samples had similar allele frequencies of each EGFR mutation. Driver mutations were found in only two ctDNA samples, both of which were mutated in EGFR, but the EGFR driver mutation in sample S4 was different in the tumour tissue and plasma. An EGFR compound mutation (L858R + S768I) was detected both in sample S8_A tissue and plasma samples. Overall, the extremely low driver mutation concordance between the FFPE and corresponding ctDNA suggested that ctDNA content in SPN patients was also too low to be efficiently sequenced by the NGS method. ddPCR was performed as a sensitive tool for low-frequency mutation testing to validate known hotspot driver mutations detected in the FFPE and ctDNA samples.


Full table
Driver mutation validation by ddPCR
ddPCR is a well-known low-frequency mutation detection platform and serves as an efficient tool to test the reliability of sequencing data from NGS. As for the limitation of ctDNA quantity, only 6 samples with EGFR/KRAS hotspot driver mutations were validated to confirm the mutation accuracy and frequency detected by NGS (Table 3). Meanwhile, four corresponding FFPE samples were also randomly selected to be validated by ddPCR as a control. A similar mutant allele frequency of FFPE samples was observed with NGS and ddPCR. Results from ddPCR detection indicated that there was a good concordance between the NGS and ddPCR detection, providing favourable evidence that the sequencing data are reliable. The ddPCR results helped prove that the mutational concordance between ctDNA and FFPE of SPN was much lower than in a previous study (16).

Full table
Malignant lung peripheral nodule prediction
A new model used to predict the malignancy of lung peripheral nodules based on the 35 plasma samples was developed using two matrixes: (I) the score of mutation (MS) contributing to the lung adenocarcinoma genesis and development and (II) the tumour burden of cfDNA (TMB), which was used to evaluate the whole mutational frequency within the panel region. Cancer gene census from the COSMIC database was used to divide the mutational genes into three groups: (I) oncogene, (II) tumour suppressor gene, and (III) non-cancer-related gene. Well-known LUAD driver mutation was added as a fourth group (such as EGFR:L858R, KRAS:G12V, ROS1/RET/ALK fusion, and so on). The score of each mutation was assigned based on the formula below:

All the potential mutational reads in the panel except germline mutations, which were identified by the normal control, were used to calculate the value:
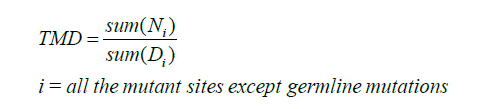
Di represents the reads of genomic i-th site; Ni represents the summary reads of non-reference base at potential mutation i.
Based on the method developed in our study, the MS and TMB values of each sample are shown in Figure 4. The green dot represents benign samples. Thus, all four benign samples were distributed in the region within TMB ≤0.2, MS ≤2. If TMB =0.3 or MS =4 was used as the cut-off value for malignant SPN prediction, 33.3% of malignant adenocarcinoma samples could be predicted accurately based on the ctDNA samples. In contrast, the sensitivity of CEA (cut-off 10 ng/mL with 97% specificity) was only ~10%, which was lower than the mutation model prediction.

Discussion
LDCT, as an imaging tool for early lung cancer screening, provided insufficient benefit to participants in this study. It is reported that 39.1% of all participants in the LDCT arm of the trial had at least one positive screen, and 96.4% of these initial positive screenings represented false positives for lung cancer (20). Overabundance of false positives could lead to higher screening costs and unnecessary invasive procedures on candidates who do not actually have lung cancer (21). According to our thousands of medical records, we found that nearly 30% of peripheral nodules in lung surgery candidates were non-malignant, and tumour serological markers do not reliably diagnose malignant nodules with high sensitivity. It seemed that protein biomarkers from serum played a less important role and produced false signals during the test. As for the non-malignant cases, some patients underwent operations because of false prediction, whereas most of the rest chose surgery out of fear of the possibility of malignancy. Thus, ctDNA is defined as a more reliable tool to deliver more specific information for both patients’ and physicians’ reference, also to further define the high-risk population, and to provide a more cost-effective method for diagnosis. ctDNA may provide an opportunity for accurate diagnosis with the advantages of non-invasiveness and no bias of heterogeneity.
Peripheral nodule DNA from surgery candidates had no significant correlation with tumour size and stage, but mutational numbers were significantly different between the benign and malignant nodules. Driver mutations were detected in 71% of malignant nodules. As for DNA mutations from the SPN plasma, advanced tumorigenesis stages and SPN size had no significant influence on somatic mutations. Moreover, the difference between benign nodules and malignant nodules was not significant. Only ~3.9% of DNA mutations from lung nodules could also be detected in the respective ctDNA by the 10,000-fold sequencing. The concordance of hotspot driver mutations between the malignant nodule DNA and the corresponding ctDNA was only 5.8%, which was much lower than the 85% concordance of cancer tissue DNA and ctDNA in the advanced tumour stage (22). Meanwhile, there was no significant difference in concordance between the stage I and those of stage II and III which were diagnosed postoperatively by incidental finding of positive lymph nodes. This might because the early ctDNA signal of peripheral nodules had not been released into the blood system, or the early DNA mutation frequency was too low to be detected with nowaday sequencing approaches. Thus, improving sensitivity of tumour detection should not be attempted through increasing depth or coverage of sequencing. Somatic mutations were significantly different between benign and malignant tissue DNA but not ctDNA, given that it could not be tested and evaluated with conservative single-gene mutations and hotspot mutations in the early tumour stage due to the possible mechanisms and pathways. Interestingly, somatic mutations were also found in benign nodules, and most of the ctDNA mutations were not detected in FFPE samples, which needed further large-scale validation study.
Mutation concordance (including driver mutation) also suggested that predicting malignant nodules through driver mutation detection based on ctDNA has limited application. This finding encouraged us to grade and score all of the specific mutations to set up a prediction model according to how strongly the mutations correlate with lung cancer. The model first integrated the whole mutational differences, which not only included ‘tumour mutational burden’ but also evaluated the influence of ‘potential mutation’. It overcame the limitation of ctDNA low-frequency mutation detection by NGS. According to this model, we could predict 33.3% of malignant patients (sensitivity) with 100% specificity. Therefore, circulating cfDNA from patients with early lung cancer could reasonably accelerate early diagnosis by ultra-deep sequencing of at least 10,000-folds depth (>1,000-fold unique reads depth) and whole tumour mutation evaluation. This model was the first non-invasive method to predict the malignancy based on ctDNA, which could benefit more than one-third of pulmonary nodule candidates. The potential clinical application of this tool, after extensive validation, is supplemental to LDCT, which yields a great number of false positive cases (7). The high specificity (100%) of the ctDNA genetic model can help us ‘rule in’ some cases (~30%) that are highly suspected to have malignant disease and should be subjected to surgery with great confidence.
More work shall be done in further studies. Because of the relatively low concordance of tissue DNA and ctDNA mutations, it was obvious that lung cancer genes-targeted capture sequencing was not efficient enough to diagnose with ideal sensitivity by simply increasing sequencing depth or coverage of ctDNA from early candidates. To achieve clinical utility, we propose that sequencing panel contents could be expanded from lung adenocarcinoma to other subtypes to better depict the performance for whole lung nodule patients. This model also shall be optimized by following larger cohort WGS sequencing data and correlated clinical data so that more cancer related gene mutations can be established in this mutational model for more sensitive differentiation in future studies. Therefore, following the remarkable findings of the cfDNA study, ctDNA could still play an important role in diagnosing nodules identified by LDCT or biomarkers as benign or malignant (21). The field is still rushing towards the identification of screening- or diagnostic-specific markers for malignant circulating cfDNA. Other techniques with theoretically higher sensitivity, such as multiplex methylation or cancer-related antibodies detection, might be incorporated to establish a multidimensional, powerful tool for early diagnosis.
Supplementary
Methods
Patient materials
A total of 1,254 consecutive candidate patients were reviewed following the IRB-approved protocols for resection of lung peripheral nodules in the First Affiliated Hospital of Guangzhou Medical University from January 2015 to November 2016. The 35 plasma and formalin-fixed, paraffin embedded (FFPE) tissue samples were collected from patients with lung peripheral solitary nodules ≤3 cm in diameter of varying size and differentiation. Complete ground glass nodules (GGNs), which were thought to be highly correlated with either non-invasive malignancies or benign changes, were not included in this study. This study is approved by ethical review board of our institution (No. 2015-25).
Blood cell/FFPE cell library preparation and NGS
The library was constructed by shearing peripheral blood cell DNA with an ultrasonoscope to generate fragments with a peak of 250 bps, followed by end repair, A-tailing, and ligation to the Illumina-indexed adapters according to the standard library construction protocol (23). Target enrichment was performed on the designed cancer-related gene capture probe (NimbleGen, Roche Sequencing, Pleasanton, CA, USA). Sequencing was performed with 2×101 bp paired-end reads and an 8-bp index read on an Illumina Hiseq 2,500/4,000 platform (San Diego, CA, USA).
ctDNA library preparation and NGS
Blood samples were collected by different hospitals in China using Cell-Free DNA BCT® blood collection tubes (Streck, La Vista, NE, USA) and transported to a clinical diagnosis lab in Tianjin. The tubes were centrifuged at 1,600 g/min for 10 min. Then, we transferred the plasma to 1.5-mL tubes and centrifuged at 18,000 g/min for 5 min to remove any remaining cells and cellular debris. Finally, we transferred the supernatant to a fresh tube and stored it at −80 °C. The ctDNA from each 2-mL volume of plasma was extracted using the QIAamp Circulating Nucleic Acid Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. We quantified the ctDNA isolated from plasma by the Qubit dsDNA HS Assay Kit (Invitrogen, Carlsbad, CA, USA). ctDNA purified from plasma was used in the subsequent NGS panel sequencing assays. The library for ctDNA was constructed with the KAPA LTP Library Preparation Kit for Illumina Platforms (Kapa Biosystems, Wilmington, MA, USA) following the manufacturer’s instructions without modification (24). Sequencing was performed with 2×101 bp paired-end reads and an 8-bp index read on an Illumina Hiseq 2,500/4,000 platform.
SNV/InDel calling
Raw reads were first processed by removing adaptors and filtering low-quality reads using SOAPnuke (http://soap.genomics.org.cn/) before aligning to the human reference GRCh37 using BWA aligner (v0.6.2-r126) (25) and removing PCR duplications by PICARD (v1.98). Then, local realignment and base quality score recalibration were performed using GATK (v2.3-9) (26). Subsequently, an in-house software was used to call candidate single nucleotide variants (SNV) using the Bayesian model, after which SNV with strand bias and read location bias were filtered using the Fisher’s exact test and Kolmogorov-Smirnov test separately (27). Then, SNVs in the local control set were filtered. SNVs were scored according to GC content, adjacent SNV and InDels, multiple mapping locations, and so on. Finally, SNVs with a low score were removed.
Candidate InDels were extracted from the CIGAR information in the BAM files. Next, the de Bruijn method was used to conduct the local de novo assemble based on the K-mers from the mapping reads (28). By comparison with the reference sequencing, InDels were predicted. InDels in the corresponding blood cell samples were removed. Finally, InDels in simple repeat regions of the human genome were checked again because of the possibility of more sequencing errors in these regions.
The method for detecting SNV/InDels in the ctDNA samples was the same as that for FFPE sequencing data, except for one additional step that was used to filter the raw mutant set. Twelve-bp paired reads were used as endogenic duplex consensus molecular barcodes and clustered (29). Those with identical barcodes and similar sequences (with consistency >80%) were considered duplication clusters of one template. The order of paired-end sequences was used to identify the sense and anti-sense strands of the template. Only the mutations with both sense and anti-sense strands were used for further analysis.
Somatic SNV and InDels were annotated by ANNOVAR, and only mutations that changed protein structure were retained for further analysis.
SV calling
Chimeric read pairs were collected and clustered to detect structural variations (SVs). The clipped parts of the soft clipped reads were collected and mapped to the genome (30). Genome locations of clipped and remaining parts were clustered to determine the accurate break points of SVs.
ddPCR
Droplet digital PCR (ddPCR, QuantStudio 3D Digital PCR System, Life Technologies, Carlsbad, CA, USA) was performed in this study. According to the guidebooks, QuantStudio 3D Digital PCR Master Mix v2 and TaqMan Assay were thawed to room temperature and mixed approximately 10 times. The targeted DNA was diluted to 200–2,000 copies/µL. The reaction mixture was prepared following the recommended protocol, and then the mixture was loaded into the QuantStudio 3D Digital PCR Chip as soon as possible.
Acknowledgements
Funding: This work was supported by National Key R & D Program of China (2016YFC0905400); Guangzhou Key Laboratory of Cancer Trans-Omics Research (GZ2012, NO348) and Guangzhou Science and Technology Project (201400000001-2 and 201400000004-5); Pearl River Nova Program of Guangzhou (No. 201506010065); project 2015B020232008 from Guangdong Province as well as National Precision Medicine Project (SQ2016YFSF090334); Chinese National Natural Science Foundation (Grant No. 81501996); Key Project of Guangzhou Scientific Research Project (Grant No. 201804020030); Guangdong Doctoral Launching Program (Grant No. 2014A030310460); and Doctoral Launching Program of Guangzhou Medical University (Grant No.2014C27); Tianjin Municipal Science and Technology Special Funds for Enterprise Development (No. 14ZXLJSY00320) and special foundation for High-level Talents of Guangdong (2016TX03R171); Key Project of Livelihood Technology of Guangzhou (2011Y2-00024).
Footnote
Conflicts of Interest: The authors have no conflicts of interest to declare.
Ethical Statement: The study protocol was reviewed and approved by the Institutional Review Board of the First Affiliated Hospital of Guangzhou Medical University (No. 2015-25). A written informed consent form, describing the purpose of the study, was signed by all of the participants.
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7-30. [Crossref] [PubMed]
- Rusch VW, Chansky K, Kindler HL, et al. The IASLC Mesothelioma Staging Project: Proposals for the M Descriptors and for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Mesothelioma. J Thorac Oncol 2016;11:2112-9. [Crossref] [PubMed]
- Kramer BS, Berg CD, Aberle DR, et al. Lung cancer screening with low-dose helical CT: results from the National Lung Screening Trial (NLST). J Med Screen 2011;18:109-11. [Crossref] [PubMed]
- Henschke CI, McCauley DI, Yankelevitz DF, et al. Early Lung Cancer Action Project: overall design and findings from baseline screening. Lancet 1999;354:99-105. [Crossref] [PubMed]
- Wahidi MM, Govert JA, Goudar RK, et al. Evidence for the treatment of patients with pulmonary nodules: when is it lung cancer?: ACCP evidence-based clinical practice guidelines (2nd edition). Chest 2007;132:94S-107S.
- Starnes SL, Reed MF, Meyer CA, et al. Can lung cancer screening by computed tomography be effective in areas with endemic histoplasmosis? J Thorac Cardiovasc Surg 2011;141:688-93. [Crossref] [PubMed]
- Bartholmai BJ, Koo CW, Johnson GB, et al. Pulmonary nodule characterization, including computer analysis and quantitative features. J Thorac Imaging 2015;30:139-56. [Crossref] [PubMed]
- Ruilong Z, Daohai X, Li G, et al. Diagnostic value of 18F-FDG-PET/CT for the evaluation of solitary pulmonary nodules: a systematic review and meta-analysis. Nucl Med Commun 2017;38:67-75. [Crossref] [PubMed]
- Fahrmann JF, Grapov D, DeFelice BC, et al. Serum phosphatidylethanolamine levels distinguish benign from malignant solitary pulmonary nodules and represent a potential diagnostic biomarker for lung cancer. Cancer Biomark 2016;16:609-17. [PubMed]
- Kupert E, Anderson M, Liu Y, et al. Plasma secretory phospholipase A2-IIa as a potential biomarker for lung cancer in patients with solitary pulmonary nodules. BMC Cancer 2011;11:513. [Crossref] [PubMed]
- Wang W, Liu M, Wang J, et al. Analysis of the discriminative methods for diagnosis of benign and malignant solitary pulmonary nodules based on serum markers. Oncol Res Treat 2014;37:740-6. [Crossref] [PubMed]
- Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 2014;20:548-54. [Crossref] [PubMed]
- Szpechcinski A, Rudzinski P, Kupis W, et al. Plasma cell-free DNA levels and integrity in patients with chest radiological findings: NSCLC versus benign lung nodules. Cancer Lett 2016;374:202-7. [Crossref] [PubMed]
- Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem 2015;61:112-23. [Crossref] [PubMed]
- Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6:224ra24. [Crossref] [PubMed]
- Izumchenko E, Chang X, Brait M, et al. Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nat Commun 2015;6:8258. [Crossref] [PubMed]
- Li S, Choi YL, Gong Z, et al. Comprehensive Characterization of Oncogenic Drivers in Asian Lung Adenocarcinoma. J Thorac Oncol 2016;11:2129-40. Erratum in: J Thorac Oncol 2017;12:408. [Crossref] [PubMed]
- Shao D, Lin Y, Liu J, et al. A targeted next-generation sequencing method for identifying clinically relevant mutation profiles in lung adenocarcinoma. Sci Rep 2016;6:22338. [Crossref] [PubMed]
- Kim EY, Cho EN, Park HS, et al. Compound EGFR mutation is frequently detected with co-mutations of actionable genes and associated with poor clinical outcome in lung adenocarcinoma. Cancer Biol Ther 2016;17:237-45. [Crossref] [PubMed]
- National Lung Screening Trial Research Team, Aberle DR, Adams AM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011;365:395-409. [Crossref] [PubMed]
- Brothers JF, Hijazi K, Mascaux C, et al. Bridging the clinical gaps: genetic, epigenetic and transcriptomic biomarkers for the early detection of lung cancer in the post-National Lung Screening Trial era. BMC Med 2013;11:168. [Crossref] [PubMed]
- Lee JY, Qing X, Xiumin W, et al. Longitudinal monitoring of EGFR mutations in plasma predicts outcomes of NSCLC patients treated with EGFR TKIs: Korean Lung Cancer Consortium (KLCC-12-02). Oncotarget 2016;7:6984-93. [PubMed]
- Head SR, Komori HK, LaMere SA, et al. Library construction for next-generation sequencing: overviews and challenges. Biotechniques 2014;56:61-4, 66, 68, passim. [Crossref] [PubMed]
- Xu S, Lou F, Wu Y, et al. Circulating tumor DNA identified by targeted sequencing in advanced-stage non-small cell lung cancer patients. Cancer Lett 2016;370:324-31. [Crossref] [PubMed]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010;26:589-95. [Crossref] [PubMed]
- McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297-303. [Crossref] [PubMed]
- Kan Z, Zheng H, Liu X, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res 2013;23:1422-33. [Crossref] [PubMed]
- Narzisi G, O'Rawe JA, Iossifov I, et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat Methods 2014;11:1033-6. [Crossref] [PubMed]
- Lanman RB, Mortimer SA, Zill OA, et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS One 2015;10:e0140712. [Crossref] [PubMed]
- Wang J, Mullighan CG, Easton J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods 2011;8:652-4. [Crossref] [PubMed]

