New molecular targeted therapies for advanced non-small-cell lung cancer
Medical Oncology Department, Hospital Universitario Puerta de Hierro Majadahonda, Madrid, Spain

|
Review Article
New molecular targeted therapies for advanced non-small-cell lung cancer
Medical Oncology Department, Hospital Universitario Puerta de Hierro Majadahonda, Madrid, Spain
|
|
Abstract
Non-small-cell lung cancer (NSCLC) is a uniformly fatal disease and most patients will present with advanced stage. Treatment outcomes remain unsatisfactory, with low long-term survival rates. Standard treatment, such as palliative chemotherapy and radiotherapy, offers a median survival not exceeding 1 year. Hence, considerable efforts have started to be made in order to identify new biological agents which may safely and effectively be administered to advanced NSCLC patients. Two cancer cell pathways in particular have been exploited, the epidermal growth factor receptor (EGFR) and the vascular endothelial growth factor receptor (VEGFR) pathways. However, novel targeted therapies that interfere with other dysregulated pathways in lung cancer are already in the clinic. This review outlines the most promising research approaches to the treatment of NSCLC, discussed according to the specific molecular pathway targeted.
Key words
advanced non-small-cell lung cancer; targeted therapies; epidermal growth factor receptor (EGFR); angiogenesis; insulin-like growth factor 1 receptor (IGF-1R); EML4-ALK fusion oncogene; proteasome inhibition; histone deacetylase inhibition; immunotherapy
J Thorac Dis 2011;3:30-56. DOI: 10.3978/j.issn.2072-1439.2010.12.03
|
|
Introduction
Lung cancer is the leading cause of cancer-related mortality worldwide in both men and women, causing approximately 1.2 million deaths per year. In the United States, in 2009, there were an estimated 219,000 new cases of lung cancer and 159,000 deaths (1). Non-small-cell lung cancer (NSCLC) accounts for > 80% of all lung cancers.
Most people diagnosed with NSCLC are unsuitable for surgery. For advanced disease, chemotherapy (CT) remains the cornerstone of treatment, with palliation and patients´ quality of life as the primary end-point. Although several advantages have been observed, the treatment of advanced fit NSCLC patients with the standard third-generation platinum-based doublet agents seems to have reached a plateau of effectiveness (2). To date, various combinations of cytotoxic drugs have not improved treatment results beyond what has been observed with platinum doublets. In contrast, major progress in the understanding of cancer biology and the mechanism of oncogenesis has allowed the development of molecular targeted therapies that block dysregulated signaling pathways and the metabolic processes that characterize lung cancer cells. These therapies achieve long-term disease control. Better toxicity profile than conventional CT, better target selectivity, availability for chronic treatment and, in some cases, oral administration have marked these targeted compounds as the most promising research drugs. Conflicting results have demonstrated marginal benefits with anti-angiogenic strategies and epidermal growth factor receptor (EGFR) inhibitors in unselected patients with NSCLC. However, oncogenic mutations in the EGFR kinase domain are strongly associated with clinical response to tyrosine kinase inhibitors (TKIs) (3). Yet even patients who are exquisitely sensitive to gefitinib or erlotinib by virtue of somatic EGFR mutations, ultimately develop resistance. So, development of additional agents that inhibit EGFR signaling in such patients remains a challenge. Furthermore, novel targeted therapies that interfere with the EML4-ALK fusion oncogene or with insulin-like growth factor 1 receptor (IGF-1R) have shown promising activity. Dysregulation in other key signalling pathways and molecules, such as PI3K/AKT/mTOR, Ras/Raf/MAPK, MET kinase or angiogenesis, have been identified as potential targets and new agents aimed at these abnormalities are being investigated. In this review we discuss the most promising targeted approaches to the treatment of NSCLC.
|
|
Second-generation EGFR TKIs
Approximately 10 to 17% of advanced NSCLC patients in Western countries harbor activating EGFR mutations (exon 19 deletions or L858R) (4), while the incidence of such mutations is higher in patients of Asian origin (3). Most of them show a dramatic initial response to treatment with the first-generation EGFR TKIs, erlotinib and gefitinib. A small number of patients with EGFR mutations have primary resistance to erlotinib and gefitinib, and most patients who initially respond to this treatment will develop acquired resistance to it. Intense research in these NSCLCs has identified two major mechanisms of resistance to EGFR TKIs: secondary resistance mutations and “oncogene kinase switch” systems. The secondary T790M mutation occurs in more than 50% of EGFR-mutated patients with TKI resistance and, in vitro, this mutation negates the hypersensitivity of activating EGFR mutations (5). Sensitive detection methods have identified a proportion of TKI-naïve tumors that carry T790M, and these resistant clones may be selected after exposure to gefitinib or erlotinib. How T790M affects hypersensitivity for activating EGFR mutations is still not completely clear. Initially, it was speculated, based on the crystallographic structure of the kinase domain of EGFR, that the bulkier methionine residue of the “gatekeeper” T790M changed the ATP-binding pocket of the kinase, thereby blocking the engagement of erlotinib or gefitinib (6). However, more recently, it was demonstrated that T790M affected binding of gefitinib to L858R-EGFR minimally. Instead, L858R-T790M-EGFR had greater affinity to ATP than L858R alone, which is predicted to decrease binding of gefitinib and erlotinib, because these drugs are ATP-competitive kinase inhibitors (7). Other secondary resistance mutations (D761Y, L747S, T854A) seem to be rare. The amplification of the MET oncogene is present in 20% of TKI-resistant tumors; however, in half the cases with this “oncogene kinase switch” mechanism, T790M is coexistent (8). It is possible that other kinases (such as IGF-1R) might also be selected to bypass EGFR pathways in resistant tumors. The problems with both primary and acquired resistance to erlotinib and gefitinib support the need for development of additional agents that inhibit EGFR signaling in such patients.
The second generation of EGFR-TKI compounds is now being introduced into clinical trials. The two most commonly employed strategies by which these drugs attempt to improve upon first-generation EGFR TKIs are introducing covalent (irreversible) binding of the drug to the drug target and broadening the affected receptor TK targets of the drug within the cell. The first-generation agents, gefitinib and erlotinib, join to their target, the catalytic site in the EGFR TK domain, through classic competitive binding with ATP. In contrast, many of the second-generation compounds form covalent and, thus, permanent bonds with their target, which should theoretically increase their effectiveness by prolonging the inhibition of EGFR signaling to the entire lifespan of the drug-bound receptor molecule. In cell culture systems, such irreversibly binding TKIs can effectively kill cells that have acquired resistance to first-generation TKIs (9). The other common theme to the design of the second-generation EGFR TKIs is kinase multi-targeting. Gefitinib and erlotinib are both fairly selective for the EGFR TK domain, while second-generation EGFR TKIs have been developed that, in addition to blocking EGFR signaling, target additional members of the ErbB family, such as Her-2 or other downstream or parallel pathways like the vascular endothelial growth factor receptor (VEGFR) pathway.
Clinical experience with the most promising second-generation irreversible EGFR TKIs is discussed below (Table 1).
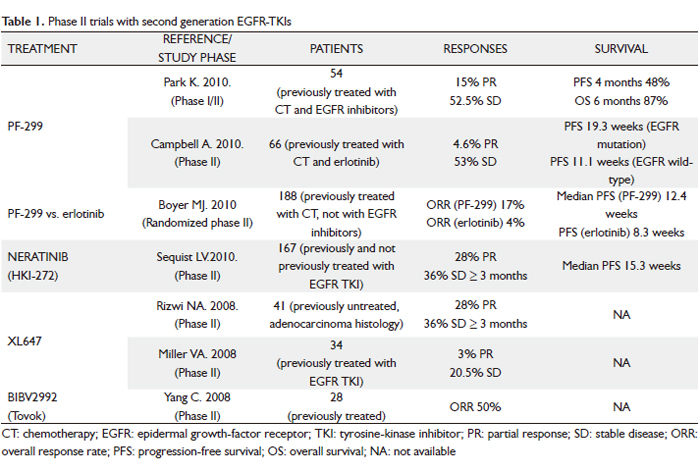 PF-00299804 (PF299)
PF-299 is an oral, irreversible, small-molecule inhibitor of the human EGFR (HER)-1, -2 and –4 tyrosine kinases, with a better in vitro and in vivo inhibition profile against EGFR-T790M than gefitinib or erlotinib (10). The drug also has activity against exon 20 EGFR insertion mutations. Furthermore, it showed adequate distribution in human tumor xenografts with optimal pharmacokinetic properties in preclinical studies. In a phase I trial of PF-299 for patients with advanced solid tumors, patients with EGFR mutations and resistance to erlotinib/gefitinib showed responses (11).
Two phase II trials have been done in patients with advanced NSCLC who had previously been treated with both CT and an EGFR inhibitor (12,13). First, an Asian study enrolled 12 patients in an initial phase I, followed by an additional 42 patients in the subsequent phase II study after a dose of 45 mg daily had been identified as the appropriate target dose (12). All patients had KRAS wild-type NSCLC (adenocarcinoma histology) refractory to platinum-based CT and erlotinib or gefitinib. The analysis presented at the 2010 American Society of Clinical Oncology (ASCO) Annual Meeting focused on the patients in the phase II portion, of whom 40 had response data available. Overall, these results were quite encouraging, with 48% of patients without progression 4 months after starting treatment, 15% showing a partial response (PR), and another 52.5% demonstrating stable disease (SD) as their best response. Overall survival (OS) rate at 6 months was 87%. Treatment was generally well tolerated. Common adverse events (AEs) were diarrhea, rash, acneiform dermatitis and mouth sores, all typically in the mild to moderate range. A similar trial of American patients was reported by Campbell and colleagues (13). A total of 66 patients with advanced NSCLC after failure of ≥ 1 CT regimen and erlotinib received PF299 45 mg once daily. Population was divided into the 50 patients with an adenocarcinoma, of whom more than half were never-smokers and the majority had an EGFR mutation, and the other 16 with a non-adenocarcinoma, who were much less likely to be never-smokers or have an EGFR mutation. Of the 62 evaluable patients, 3 had a PR and 35 SD ≥ 6 weeks. Authors saw a wide range in duration of treatment, with 13 patients (23%) demonstrating prolonged clinical benefit (either a complete response [CR], PR or SD lasting at least 24 weeks). Not all these patients had an EGFR mutation (7 did, 4 were EGFR wild-type and 2 were unknown). Overall, progression-free survival (PFS) was longer in patients who had an EGFR mutation (n = 26, median PFS 19.3 weeks) than in those with EGFR wild-type (n = 20, median PFS 11.1 weeks). The toxicity profile was similar to the Asian experience, though the investigators noted that the side effects seemed to gradually decrease with ongoing treatment. Common AEs included diarrhea, fatigue, rash and stomatitis/mucosal inflammation, mainly grade 1/2 and manageable. Overall, both these studies show that an encouraging fraction of extensively treated patients with advanced NSCLC have a PR or at least prolonged SD with this oral agent, and that the results, while perhaps most encouraging in patients with an EGFR mutation or the demographic features where it is prevalent, convincingly reveal activity in patients who do not have an EGFR mutation. A phase III trial (BR 26) is studying PF-299 compared with placebo in treating patients with advanced NSCLC that has not responded to previous EGFR TKI therapy for advanced stage.
Another way to ask how much PF299 offers over and above a currently available standard oral EGFR inhibitor is to compare the two directly in patients who have never received prior therapy against EGFR. Boyer et al. conducted a randomized phase II trial, which was also presented at the 2010 ASCO Annual Meeting, to answer this question (14). The study enrolled 188 patients with advanced NSCLC and tissue available for molecular marker studies, who had previously received one or two prior lines of CT but had not received EGFR inhibitor therapy. Patients were randomized 1:1 to receive oral PF299 45 mg or erlotinib at the standard dose of 150 mg once daily until progression or toxicity. The primary end-point was PFS. Overall, baseline characteristics were well balanced, but the PF299 arm had a higher proportion of patients with an EGFR mutation (20% vs 12%). On the other hand, more patients on the PF299 arm had a marginal performance status of 2 (19% vs 3%), which would be expected to disfavor it. Mutation status determination rates were high (KRAS = 81%; EGFR = 77%). The trial demonstrated significantly higher overall response rate (ORR) with PF299 vs erlotinib (17% vs 4%; P=0.009), as well as significantly improved PFS in patients treated with PF299 in the overall trial population (HR = 0.68; 95% CI, 0.49-0.95; P=0.019). What was especially interesting was that in all subgroups (EGFR mutation or wild-type, KRAS mutation or wild-type, adenocarcinoma or non-adenocarcinoma, men or women, never-smoker or ever-smoker) the trend favored PF299 to a similar degree of about 30-40%. Common EGFR TKI AEs were more frequent with PF299. Acneiform dermatitis, as opposed to rash, was more common with PF299. Also, paronychia, mouth sores and diarrhea were all more common and tended to be more severe with PF299 vs erlotinib. Eight patients (6 in the PF299 arm) discontinued due to AEs. The authors concluded that this line of research was promising enough to warrant a larger randomized phase III trial with the same design.
Neratinib (HKI-272)
Neratinib is an irreversible pan-ErbB TKI (EGFR, ErbB2, ErbB3) with in vitro and in vivo activity against EGFR mutations (L858R, exon 19 deletions), exon 20 EGFR insertions (which are more resistant to gefitinib/erlotinib), amplified or mutated ErbB2, and compound EGFR mutations with T790M. In a phase I dose-escalation trial including 73 patients whose tumors expressed either the EGFR or Her-2 receptor, neratinib was shown to be well tolerated, with primary toxicities of diarrhea, nausea, asthenia, and anorexia (15). The dose-limiting toxicity was grade 3 diarrhea at 400 mg/day, establishing the maximum-tolerated dose (MTD) as 320 mg/day. Twelve patients with NSCLC were enrolled. Despite no responses noted in the NSCLC cohort, 5 patients with acquired resistance to gefitinib/erlotinib had SD for more than 24 weeks. These findings led to a 3-arm randomized phase II trial of neratinib in which a total of 167 patients were divided into 3 groups: arm A, progression after >12 weeks of erlotinib or gefitinib treatment and tumor positive for EGFR mutation (n = 91); arm B, progression after >12 weeks of erlotinib or gefitinib treatment and tumor negative for EGFR mutation (n = 48); and arm C, no prior EGFR TKI treatment, adenocarcinoma, 16). All patients received daily oral neratinib, initially at 320 mg but subsequently reduced to 240 mg because of excessive diarrhea. The primary end-point was ORR. Diarrhea was the most common toxicity; grade 3 incidence was 50% at 320 mg, but improved to 25% after dose reduction. The activity of neratinib was low in all patients tested. The RR was 3% in arm A and zero in arms B and C. No patients with known T790M responded. However, molecular analysis revealed a striking 75% RR among the four patients with the rare EGFR mutation G719X (where X indicates the substitution of the glycine residue for another, typically serine, cysteine or alanine). G719Xcomprises <5% of EGFR mutations and has been associated with sensitivity to gefitinib and erlotinib. Preclinical models comparing the relative sensitivity of various EGFR mutations to erlotinib and neratinib have demonstrated that erlotinib may be more selective at inhibiting exon 19 deletion mutations, and neratinib may be more effective for point mutations, including those at codon G719 (17). Interestingly, although the distribution of exon 19 deletion mutations and L858R is typically equal at diagnosis (18), in the study population of this trial the distribution of mutations was three to one in favor of exon 19 deletions. This may have also contributed to the low observed neratinib activity because, as mentioned, preclinical data suggest that point mutations like L858R are more readily inhibited by neratinib than exon 19 deletions (17). On the other hand, lowering the dose for excesive diarrhea may have decreased drug bioavailability below the threshold for most EGFR mutations. The prior phase I study found the average maximum concentration of neratinib after a daily dose of 240 mg was 73.5 nm/ml, which corresponds to 131 nmol/L (15). Though not directly measured on our study, the typical steady-state neratinib concentration at 240 mg daily may have been at or below the 60 nmol/L required to inhibit the E746-A750 exon 19 deletion on the 90 to 800 nmol/L required to inhibit T790M (based on preclinical models) (17,19). In contrast, the inhibitory concentration of the G719S mutation (3nmol/L) was likely readily achieved. In conclusion, future studies with neratinib in NSCLC will focus on attempt to modify the dose and/or schedule to mitigate diarrhea and allow for achievement of higher biologic doses. Moreover, these data highlight the importance of obtaining comprehensive genetic information on trials examining strategies for treating acquired resistance to EGFR TKIs. Development of noninvasive analysis, such as circulating tumor cell-based strategies, will facilitate this.
XL647
XL647 is an orally bioavailable reversible small molecule inhibitor of multiple receptor TK involved in tumorigenesis and angiogenesis, including EGFR, HER2, VEGFR-2 and EphB4, among other kinases. In an EGFR L858R-T790M–mutated model (H1975), XL647 was able to inhibit the growth of tumors at a lower concentration than that achieved by gefitinib or erlotinib. A phase I study of 31 patients with advanced solid tumors showed that XL647 is well tolerated at doses of up 300 mg daily (20). In a phase II trial of an enriched NSCLC population with CT-naive EGFR-mutated tumors, XL647 had activity against classic (L858R, exon 19 deletions) EGFR mutations, showing a 28% PR rate and 36% SD for ≥ 3 months (21). These findings led to a phase II trial of XL647 at 300 mg/day in patients with relapsed or recurrent NSCLC after clinical benefit with erlotinib/gefitinib for over 3 months before progression or the T790M mutation (22). The trial tested for T790M in the plasma of all patients. Accrual was complete, and preliminary results were presented at the 2008 ASCO Annual Meeting. Of the estimated 34 patients enrolled, only 1 achieved a PR and 7 had SD at their first assessment. None of the patients with T790M reported achieved a radiographic response, and most of the patients had PD within the first 2 months of drug use in the study. Final results of the study are awaited.
BIBW 2992 (Tovok)
BIBW 2992 is another potent, oral, irreversible TKI of EGFR and ErbB2. In vitro and in vivo models of EGFR-mutated NSCLC have shown that BIBW 2992 might inhibit EGFR-mutated tumors with lower concentrations than neratinib. Specifically, BIBW 2992 was able to shrink tumors in transgenic mice with the L858R-T790M mutation and was effective in exon 20 insertion EGFR mutations (23). In a phase II trial of 28 patients with EGFR mutations, BIBW 2992 led to responses in 12 out of 24 evaluable patients with EGFR exon 19 deletion, L858R, L861Q and G719S/S768I mutations (50%, 95% CI, 30-70%) (24). Manageable cutaneous toxicity and diarrhea are the main adverse events. These data have led to the launch of a clinical trial of BIBW 2992 at 50 mg/day as third-line therapy for NSCLC patients who have failed CT and had previous clinical benefit from erlotinib/gefitinib before progression. Another phase IIb/III randomized trial of BIBW 2992 50 mg once daily versus placebo plus best supportive care in patients with NSCLC, who had received previous treatment with at least one but not more than two lines of cytotoxic CT (one line must have been a platinum-containing regimen) and either gefitinib or erlotinib (LUX-Lung 1), is ongoing. An unblinded interim analysis of tumor response and safety by an independent Data Monitoring Committee after the first evaluable patients treated with BIBW 2992 determined continuation for full accrual (25). Finally, BIBW2992 is also being evaluated as first-line treatment and in previously treated patients in combination with both CT and targeted agents (sirolimus, cetuximab).
Final results of the above-mentioned trials will help determine whether BIBW 2992 has clinical activity against EGFR-mutated tumors with acquired resistance to erlotinib/gefitinib.
|
|
Multi-targeted TKIs
In addition to erlotinib and gefitinib, which specifically target the EGFR pathway, efforts to identify drugs that inhibit key pathways involved in the pathogenesis of NSCLC have led to the development of multi-targeted agents. Small-molecule TKIs that inhibit receptors such as VEGFR-2, EGFR, PDGFR, Raf and KIT simultaneously may offer advantages over agents with single targets, and therefore a higher likelihood of single-agent activity. In addition, because multi-targeted TKIs are often available orally, they may be more convenient for patients. However, a potential disadvantage is the possible toxicity of off-target kinase inhibition and the additional toxicity when the agents are combined with CT, which may be particularly relevant. Clinical experience with these agents is described below (Table 2).
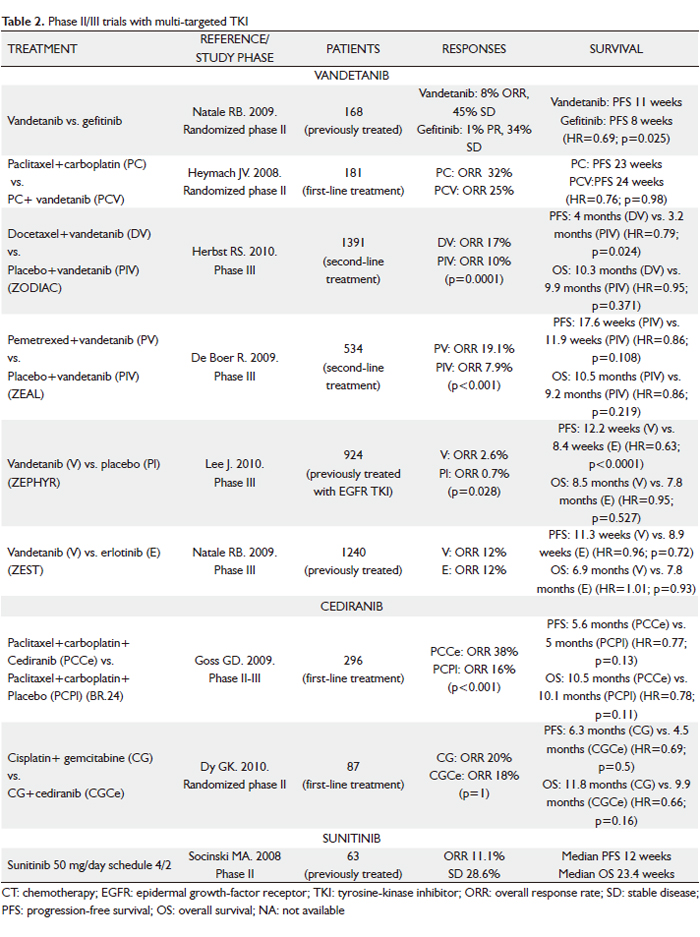  Vandetanib
Vandetanib (ZD6474; Zactima) is an oral ATP mimetic small molecule that inhibits VEGFR-2, EGFR and Rearranged during Transfection (RET) TK. Phase I dose-escalation studies in American/Australian and Japanese patients with a broad range of advanced tumors demonstrated that vandetanib monotherapy was generally well tolerated at daily oral doses up to 300 mg, and its half-life of approximately 120 hours support a once-daily dosing (26,27).
Vandetanib at a dose of 300 mg daily was compared to gefitinib in a randomized phase II study with 168 previously treated NSCLC patients (28). PFS, the primary end-point, was longer in those receiving vandetanib (11 weeks vs 8 weeks; hazard ratio [HR]=0.69; 95% CI, 0.50-0.96; P=0.025). At the time of disease progression, patients were allowed to cross over, which may explain why there was no significance difference in OS between the treatment arms. Two other phase II randomized trials assessed the efficacy of vandetanib in combination with standard CT (29,30). In first-line treatment, vandetanib, as a monotherapy or in combination with paclitaxel and carboplatin, was compared with paclitaxel and carboplatin in 181 chemo-naïve patients with advanced NSCLC (31). The vandetanib monotherapy arm was stopped early since it was less effective than CT. No difference was observed between the two remaining treatment arms in terms of survival. In second-line treatment, a total of 127 patients with metastatic NSCLC after failure of first-line platinum-based therapy were randomized to receive vandetanib (100 or 300 mg/day) plus docetaxel (75 mg/m2 every 21 days) or placebo plus docetaxel (30). Interestingly, the lower dose level resulted in an improvement in PFS when added to docetaxel (18.7 weeks vs 12 weeks with docetaxel alone; HR=0.64; P=0.037), whereas the higher dose did not result in a significant improvement when combined with docetaxel (17 weeks; HR=0.83; P=0.231). However, there was no statistically significant difference in OS among three arms. One explanation for the lack of PFS benefit at the higher dose level is possible antagonistic effects with increasing inhibition of EGFR when used concurrently with CT. Certainly, earlier studies did not suggest any benefit from use of EGFR TKIs with CT in unselected patients.
The promising results of these trials led to the evaluation of vandetanib as second-line treatment of patients with locally advanced or metastatic NSCLC in several large phase III studies (31-34). Two randomized, placebo-controlled clinical trials have investigated the efficacy of the addition of vandetanib to CT: the Zactima in cOmbination with Docetaxel In non-small cell lung Cancer (ZODIAC) trial (31), comparing vandetanib (100 mg) plus docetaxel with docetaxel monotherapy (75 mg/m2 intravenously every 21 days; maximum six cycles) in 1391 patients; and the Zactima Efficacy with Alimta in Lung cancer (ZEAL) trial (32), comparing vandetanib (100 mg/day) and 500 mg/m2 pemetrexed every 21days (maximum 6 cycles) with pemetrexed monotherapy in 534 patients previously treated with one prior anticancer therapy for advanced NSCLC. In the ZODIAC study (31), vandetanib significantly prolonged PFS, the primary objective of the study, compared to placebo (median 4 vs 3.2 months; HR 0.79; 95% CI 0.70-0.90; P<0.0001),although there was no statistically significant difference in OS (median survival 10.3 vs 9.9 months; HR 0.95; 95% CI 0.84-1.07; P=0.196). Preplanned subanalysis showed efficacy across most of the subgroups (male vs female; smokers vs non-smokers; adenocarcinoma vs squamous-cell carcinoma). In the ZEAL study (32), a smaller, less “well-powered” (to show a statistically significant difference) trial, similar scales of improvement in PFS (HR 0.86; 95% CI 0.69-1.06; P=0.108) and OS (HR 0.86; 95% CI 0.65-1.13; P=0.219) were observed in patients given vandetanib compared to placebo, but these were not statistically significant. There were statistically significant advantages for ORR (19.1% vs 7.9%; P<0.001) and time to deterioration of symptoms (TDS) (HR 0.61; P=0.004).
Two further randomized trials have evaluated the efficacy of vandetanib as a single agent: the Zactima Efficacy trial for NSCLC Patients with History of EGFR-TKI chemo-Resistance (ZEPHYR) trial (33), which tested 300 mg/day vandetanib vs placebo in a refractory population who failed CT and anti-EGFR therapy; and the Zactima Efficacy when Studied vs Tarceva (ZEST) trial (34), which compared 300 mg/day vandetanib with erlotinib in 1240 patients with advanced NSCLC after failure of at least one prior anticancer therapy.
The ZEPHYR study (33) did not meet its primary objective of demonstrating an OS benefit with vandetanib vs placebo (median OS 8.5 vs 7.8 months; HR=0.95; 95% CI 0.81-1.11; P=0.527), although PFS (HR=0.63; 95% CI, 0.54-0.74; P<0.0001) and ORR (2.6% vs 0.7%; P=0.028) were significantly better. There was no difference in PFS with vandetanib vs erlotinib (HR=0.98; 95% CI, 0.87-1.10; P=0.721) in the ZEST trial (34). However, in a preplanned non-inferiority analysis, vandetanib and erlotinib showed equivalent efficacy for PFS and OS.
Cediranib
Cediranib (AZD2171; Recentin), an oral small-molecule inhibitor of the TK domain of all three VEGFR, VEGFR-1, VEGFR-2 and VEGFR-3, as well as the TKs associated with platelet derived growth factor (PDGF) receptors, demonstrated activity in preclinical models and phase I trials (35,36). A phase I study showed encouraging tumor control with the use of cediranib at doses of 30 mg and 45 mg in addition to carboplatin/paclitaxel in patients with advanced NSCLC (35). Nine of the 20 enrolled patients had PR, while 11 had SD. Both doses appeared tolerable and equally active. A second trial evaluated cediranib (30 mg and 45 mg) in combination with cisplatin/gemcitabine in advanced NSCLC (36). Central review confirmed responses in 4 of 15 patients in this trial and a further seven had SD. Both trials concluded that the recommended phase II dose of cediranib in combination with CT was 45 mg daily.
On the basis of these data, a randomized phase II-III trial comparing cediranib with placebo in conjunction with paclitaxel plus carboplatin in advanced (stage IIIB/IV) NSCLC was performed (BR24 Study) (37). A total of 296 patients were enrolled. First, 45 patients received 45 mg/day of cediranib, but, after deaths related to toxicity in the cediranib arm (hemoptysis, febrile neutropenia, diarrhea, atrial fibrillation, cerebral embolism), the protocol was amended to reduce the doses of cediranib from 45 to 30 mg and to limit accrual to patients with good performance status. In early 2008, Astra Zeneca reported that the study would not continue into phase III following the planned end of phase II efficacy and tolerability analyses. In the primary phase II analysis (30 mg cohort), the addition of cediranib to carboplatin/paclitaxel resulted in improved ORR (38% vs 16%; P<0.0001) and PFS (5.6 months for cediranib and 5 months for placebo; HR=0.77; 95% CI, 0.56-1.08; P=0.13). However, the cediranib-based combination was associated with excess toxicity (severe hypertension, hypothyroidism, hand-foot syndrome, diarrhea, febrile neutropenia, hemoptysis); hypoalbuminemia, age > 65 years and female sex predicted increased toxicity. Consequently, a reduced dose of cediranib (20 mg/day) plus carboplatin and paclitaxel will be investigated in a randomized trial in patients with a good performance status, no significant weight loss and no hypoalbuminemia.
A second phase II study, recently presented at the 2010 ASCO Annual Meeting, evaluated the safety and efficacy of cediranib, with carboplatin and gemcitabine as first-line therapy for advanced NSCLC (38). Eighty-seven patients were randomized 2:1 to carboplatin-gemcitabine+cediranib at 30 mg/day (arm A) vs carboplatin-gemcitabine alone (arm B). The trial did not meet its primary end-point of ORR (the ORR in arm A and B was 20% vs 18%; P=1.0). However, the secondary end-points of PFS at 6 months were met, with a trend towards improved PFS (median PFS of 6.3 vs 4.5 months; HR=0.69; P=0.15) and OS (median OS of 11.8 vs 9.9 months; HR=0.66; P=0.16) when cediranib was added to carboplatin-gemcitabine. Treatment was generally well tolerated, with patients in arm A experiencing more grade >3 non-hematologic AEs.
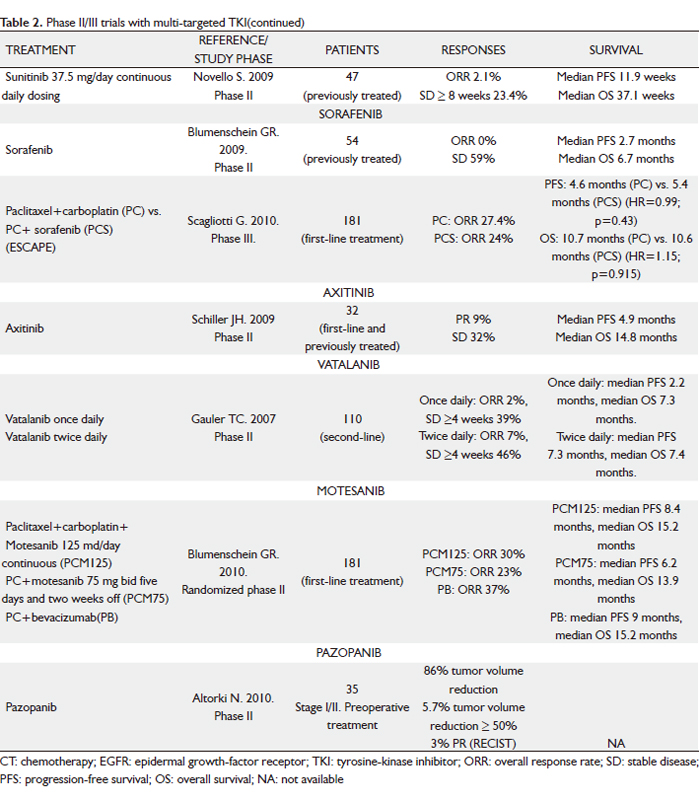
Sunitinib
Sunitinib malate (SU11248; Sutent) is an oral, selective multi-targeted TK inhibitor with anti-angiogenic and antitumor activities. It inhibits VEGFR-1, -2 and –3 and PDGFR-α and –β activity, as well as the activity of several related TKs (KIT, fms-like TK receptor 3 [FLT3], CSF-1R and RET). In preclinical studies, sunitinib effectively inhibited the growth of established human NSCLC xenografts; antitumor activity was also observed in patients with NSCLC in a phase I study of sunitinib plus gemcitabine and cisplatin (39). A phase II trial of sunitinib for previously treated advanced NSCLC investigated two dose schedules (40,41). The first dosing schedule studied was the standard 4 weeks on and 2 weeks off regimen (50 mg/day for 4 weeks followed by 2 weeks of no treatment in 6-week treatment cycles) (40). Of the 63 patients included, seven had confirmed PR, yielding an ORR of 11.1% (95% CI, 4.6 to 21.6%), similar to the currently approved agents despite the evaluation of sunitinib in a more heavily pre-treated patient population (60% of patients had received two or more prior systemic treatment regimens). An additional 18 patients (28.6%) experienced SD of at least 8 weeks. Median PFS was 12 weeks and median OS was 23.4 weeks. Treatment was generally well tolerated, with the majority of AEs being grade 1 or 2. The most commonly reported AEs were asthenia, pain/myalgia, nausea/vomiting and stomatitis. Two fatal pulmonary hemorrhages were reported (one assessed as drug-related), both in patients with squamous cell histology. An additional drug-related death from a cerebral hemorrhage was described. The second dosing schedule consisted of continuous daily sunitinib at a lower dose (37.5 mg) (41). Following reports from a phase II sunitinib study in metastatic breast cancer, suggesting that some patients had increases in the size of surface lesions during the 2-week off-treatment period (42), it was hypothesized that better tumor control could be achieved with sunitinib given once daily on a continuous daily dose (CDD) schedule. Despite relatively low ORR of 2.1%, 11 (23.4%) patients had SD > 8 weeks and five had SD > 6 months. It is noteworthy that the median PFS (11.9 weeks) and median OS (37.1 weeks) are comparable to the currently available treatment options for this setting, as shown in phase III studies, including docetaxel, erlotinib and pemetrexed vs docetaxel. The sunitinib AE profile observed in the CDD cohort was tolerable and manageable. Although formal comparisons cannot be made between the safety profiles of sunitinib on schedules 4/2 vs CDD, among the most commonly reported toxicities, constitutional (e.g. fatigue/asthenia: 69.8 vs 59.6%) and gastrointestinal (e.g. nausea/vomitng: 52.4 vs 40.4%) AEs appeared to be less frequent on the CDD schedule, despite its longer median treatment duration.
Sunitinib at a starting dose of 37.5 mg with continuous daily dosing was also assessed in 64 NSCLC patients who had received whole brain radiation therapy for brain metastases and 43). Antitumor efficacy was based on overall (RECIST) and intracranial (WHO criteria) tumor assessments. SD was reported in 16 (27%) of 60 patients via RECIST and in 6 (26%) of 23 patients with measurable brain metastases via WHO; one patient (4%) had an intracraneal PR. Median PFS was 9.4 weeks and median OS was 26 weeks. Toxicity was generally manageable and no cases of cerebral hemorrhage were reported.
Sorafenib
Sorafenib (BAY 43-9006; Nexavar) is an oral multikinase inhibitor that inhibits the serine threonine kinases, c-Raf and b-Raf; the TK receptors VEGFR-1, -2 and –3 and PDGFR β; FLT3, the proto-oncogen RET and c-KIT. This activity profile allows sorafenib to slow tumor growth directly by inhibiting the Ras/Raf/MEK/ERK signaling pathway and indirectly by targeting tumor vasculature and angiogenesis. In phase I and phase II studies in patients with NSCLC, single-agent sorafenib demonstrated antitumor activity (44-47). In a phase II study of patients with advanced, previously untreated NSCLC, an ORR of 12% was observed in sorafenib-treated patients, and the median OS was 8.8 months (45). Sorafenib was also active in patients with recurrent NSCLC. In a randomized, discontinuation, phase II study (ECOG 2501) of sorafenib vs placebo in patients with NSCLC in whom at least two prior CT regimens had failed, patients were randomly assigned to receive either sorafenib or placebo after receiving sorafenib for 8 weeks and having SD when evaluated for response (46). Those randomly assigned to receive sorafenib had a significantly greater disease control rate (DCR) (as measured by CR plus PR plus SD) (47% vs 19%; P=0.01) and median PFS (3.6 vs 1.9 months; P=0.01) than those who received placebo. Another phase II trial involving 52 advanced NSCLC patients who received one or two prior systemic therapies reported a 59% SD rate (47). Although no responses per standard size criteria were noted (30% decrease in the sum of the longest diameter of target lesions), tumor shrinkage or cavitation was observed in 29% of patients. Median PFS was 2.7 months; and median OS was 6.7 months. Patients with SD had a median PFS of 5.5 months. Major grade 3 to 4 treatment-related toxicities included hand-foot skin reaction (10%), hypertension (4%), fatigue (2%) and diarrhea (2%). These results question the utility of anatomic responses by RECIST for evaluating molecular-targeted therapies. The RECIST criteria were developed as a surrogate end point for the efficacy of cytotoxic agents. Sorafenib has a multimodal mechanism of action that results in inhibition of both angiogenesis and tumor growth, two characteristics not easily measured by RECIST. Sorafenib monotherapy continues to be evaluated as a third- and fourth-line treatment in patients with advanced NSCLC in a large phase III study.
On the basis of promising clinical activity observed in two phase I trials of sorafenib plus carboplatin and paclitaxel, with a total of 26 evaluable patients (48,49), a large, multicenter, randomized, placebo-controlled, phase III study - the ESCAPE (Evaluation of Sorafenib, Carboplatin and Paclitaxel Efficacy in NSCLC) trial – was run (50). A total of 926 patients were randomly assigned to receive up to six 21-day cycles of carboplatin area under the curve 6 and 200 mg/m2 paclitaxel (CP) on day 1, followed by either 400 mg sorafenib twice a day (arm A) or placebo (arm B). The maintenance phase after CP consisted of 400 mg sorafenib or placebo twice a day. The primary end-point was OS. On the basis of a planned interim analysis, median OS was 10.7 months in arm A and 10.6 months in arm B (HR=1.15; P=0.915). The study was terminated early after the interim analysis concluded that it was highly unlikely to meet its primary end-point. In a pre-specified exploratory analysis of patients with squamous histology (24% of the patient population), those on the sorafenib-containing arm compared with the CT-alone arm experienced a shorter PFS (median PFS of 4.3 and 5.8 months, respectively; HR=1.31; 95% CI, 0.94-1.83) and a statistically significant shorter OS (median OS of 8.9 and 13.6 months, respectively; HR=1.85; 95% CI, 1.22-2.8). Patients with other histologies had similar PFS and OS in the two treatment arms. The rate of grade > 3 pulmonary hemorrhage was 1.1% in each arm (n=5); and drug-related deaths were observed in 13 patients (2.8%) in the sorafenib-containing arm and 4 patients (0.9%) in the CT-alone arm. Thus, it does not appear that excessive toxicity contributed to the worse survival among patients with squamous histology in the sorafenib-containing arm. Squamous histology was associated with a higher rate of fatal bleeding, irrespective of treatment arm.
The findings of the ESCAPE trial (50) follow the challenging history of many targeted agents in combination with CT in NSCLC and question the method of developing these drugs. Several molecular therapies, including erlotinib (51), gefitinib (52) or cediranib (37) showed promising results in combination with CT in early drug development that could not be confirmed in subsequent randomized, placebo-controlled, phase III trials. Several factors may contribute to negative results, including the choice of platinum-doublet regimen, the inclusion of patients with squamous cell carcinoma, or specific disease characteristics, such as a specific biomarkers. The backbone carboplatin-paclitaxel CT used in this trial was recently evaluated with sorafenib in refractory advanced melanoma with disappointing results (53), leading to speculation that sorafenib could alter the pharmacokinetics of carboplatin-paclitaxel, thereby impairing the efficacy of the combined regimen compared with carboplatin-paclitaxel alone. The basis for the phase III trial by Scagliotti et al (50) was the rates of response and stable disease observed in previously described phase I trials (48,49). However, the inherent flaws of response as an end point, the small sample size, and lack of a control arm make the interpretation of the efficacy data from these trials difficult. In retrospect, a well-designed randomized phase II trial might have provided additional efficacy and toxicity data among patients with squamous and nonsquamous histology and might have assisted in the optimal development of a phase III trial. On the other hand, the effective use of VEGFR TKIs will depend on identifying patients who are most likely to benefit from therapy. Similar to what we learned from EGFR mutations testing, only standardized and validated molecular assessment along with a precise understanding of disease biology is likely to provide reliable information for making rational clinical decisions. Unfortunately, there are no proven biomarkers for selecting patients with NSCLC who would benefit from antiangiogenic therapy, despite active research and a bounty of candidate markers.
Axitinib
Axitinib (AG-013736) is an oral, potent, selective inhibitor of VEGFR-1, -2 and –3, and a relative of most other VEGFR-TKIs at clinical doses. It is currently being studied in multiple solid tumors. In a phase II study 32 patients with advanced NSCLC were treated with single-agent axitinib (in the first-line, second-line or third-line setting) (54). Axitinib was administered at a starting dose of 5 mg orally twice daily. The dose could be escalated in 2-mg increments up to a maximum of 10 mg twice daily if no treatment-related AEs of grade >3 occurred for 2 weeks. Intrapatient dose escalations were not permitted if blood pressure was more than 150/90 mmHg or the patient was receiving medication for hypertension. Three patients (9%) had a RECIST PR and DCR was 41%. Median PFS of 4.9 months and median OS of 14.8 months are encouraging, comparing favorably with recent phase II reports evaluating monotherapy with other TKIs in similar patient populations (40,45). One-year survival rates for patients with or without prior therapy for metastatic disease were 57% and 78%, respectively. Treatment was generally well tolerated. Grade 3 treatment-related AEs in > 5% of patients comprised fatigue (22%), hypertension (9%) and hyponatremia (95%).
Vatalanib
Vatalanib (PTK787) is an oral anti-angiogenic compound blocking all currently known VEGF receptors (VEGFR 1-3), as well as PDGFR and KIT, which is currently being studied in phase II/III trials. Data from a phase II study examining the efficacy and safety of vatalanib in pre-treated patients with advanced NSCLC have been reported (55). Fifty-five patients received a fixed dose of 1,250 mg PTK787 once daily or twice daily (500 mg a.m. + 750 mg p.m.) for continuous treatment until disease progression or unacceptable toxicities. Treatment appeared active, with a trend toward greater efficacy with twice-daily treatment (11% of evaluable patients had a PR in this cohort). PFS/OS were 2.4/7.0 months for the once daily and 3.7/6.8 months for the twice daily cohort. Treatment was well tolerated, with no apparent differences between once- and twice-daily dosing.
Motesanib
Motesanib (AMG 706) is a small-molecule antagonist of VEGFR-1, -2 and –3, PDGFR, KIT and RET, which is currently in clinical development in multiple tumor types, including NSCLC. A phase Ib study showed that treatment with motesanib was tolerable when combined with carboplatin/paclitaxel and/or panitumumab, with little effect on motesanib pharmacokinetics at the 125-mg once-daily dose level (56). Treatment-related AEs were generally mild to moderate, with fatigue and hypertension as the most common grade 3 AEs. A randomized, phase II study of motesanib or bevacizumab in combination with paclitaxel and carboplatin for advanced non-squamous NSCLC was reported at the 2010 ASCO Annual Meeting (57). Patients (n=181) were randomized (1:1:1) to receive paclitaxel (200 mg/m2) + carboplatin (AUC6 mg/ml/min) on day 1 of each 3-week cycle (6 cycles maximum) plus motesanib orally at either 125 mg once daily continuously (arm A) or 75 mg twice daily for 5 days, followed by 2 treatment-free days (arm B), or paclitaxel/carboplatin + bevacizumab 15 mg/Kg once every 3 weeks on day 1 of each cycle (arm C) until disease progression or intolerability. Authors concluded that the estimated efficacy of 125 mg motesanib once daily continuously + paclitaxel/carboplatin (ORR 30%, median PFS 8.4 months, median OS 15.2 months) was similar to bevacizumab +paclitaxel/carboplatin (ORR 37%, median PFS 9 months, median OS 15.2 months). Motesanib twice daily dosing had relatively lower efficacy than the other arms. The toxicity profile was adequate.
On the basis of previously reported data, a phase III trial (MONET1) to determine if treatment with motesanib (125 mg daily) in combination with paclitaxel and carboplatin improves OS, compared to treatment with placebo in combination with paclitaxel and carboplatin, is ongoing. It was temporarily closed because of a higher risk of hemoptysis in patients with squamous cell histology. These patients were discontinued and the study was re-started; patients with non-squamous NSCLC (approximately two-thirds of the original study population) are continuing on treatment or to be enrolled.
Pazopanib
Pazopanib (GW786034) is a selective, orally available, small molecule inhibitor of VEGFR-1, -2 and -3, PDGF-α, PDGF-β and c-kit TK, which is currently in phase II development in advanced NSCLC. In a recent phase I study, pazopanib was generally well tolerated and demonstrated antitumor activity across various tumor types (58). A monotherapy daily dose of 800 mg was selected for phase II studies. In a phase II trial, short-term preoperative pazopanib at a dose of 800 mg/day demonstrated antitumor activity in patients with early-stage (stage I/II) NSCLC (59). Thirty patients (86%) achieved tumor-volume reduction after pazopanib treatment, two patients achieved tumor-volume reduction >50%, and three patients had PR according to RECIST criteria. The tolerability profile in this setting was favorable. The most common AEs included grade 2 hypertension, diarrhea and fatigue. Several pazopanib target genes and other angiogenic factors (PDGFR-α, PDGFR-β, VEGFR-1, VEGFR-2 and VEGF-C) were significantly induced in the treated tumor samples, although no statistically significant association was found between changes in gene transcript levels and tumor volumetric change, probably due to the limited sample size in this study. Various phase II studies of pazopanib in patients with advanced NSCLC have either been completed or are ongoing. These incude pazopanib monotherapy in previously treated patients, as well as combination studies with paclitaxel or pemetrexed in first-line treatment. In addition, a randomized phase II study of erlotinib plus pazopanib vs erlotinib plus placebo in second- or third-line treatment is currently recruiting participants.
BIBF 1120
BIBF 1120 (Vargatef™) is an oral triple angiokinase inhibitor that inhibits VEGFR-2, PDGFR and fibroblast growth factor receptor (FGFR). Phase I data in patients with solid tumors established the phase II dose as 200 mg twice daily and showed that the toxicities at this dose were manageable (60). Results from a phase II trial of BIBF 1120 involving patients with advanced NSCLC were reported at the 13th World Conference on Lung Cancer (61). This double-blind multicenter trial included patients with an ECOG performance status score of 0-2 who had relapsed following the failure of first- or second-line CT and showed that BIBF 1120 has single-agent activity in this population. AEs were most often gastrointestinal: the most common grade 1-3 toxicities were vomiting, nausea, diarrhea, anorexia and abdominal pain. Of particular note were results from a subset of 57 patients with ECOG-performance status of 0 or 1: these patients experienced a higher SD rate of 59%, longer PFS (median PFS was 2.9 months) and longer OS (median OS was 9.5 months) than the overall study population.
|
|
Insulin-like growth factor receptor (IGF-R) inhibitors
The insulin-like growth factor 1 (IGF-1) receptor (IGF-1R) and its ligands play a key role in lung cancer. At the cell level, after the binding of IGFs to the receptors, conformational changes in the IGF-1R result in the activation of its TK domain, in the phosphorylation of insulin receptor substrate proteins and in the activation of various intracellular signaling pathways, including the RAS/RAF/MAP kinase and the PI3K pathways. The activation of these pathways led to oncogenic transformation growth and survival of cancer cells (62).
Multiple lines of evidence suggest involvement of the IGF pathway across a range of malignancies, including NSCLC (63). Elevated plasma levels of IGF-1 have been associated with an increased risk of lung cancer; and high plasma levels of insulin-like growth factor binding protein-3 (IGFBP3) have been associated with a reduced risk. Similarly, IGFBP3, promoter of methylation in tumor cells, has been linked to decreased survival in stage I NSCLC patients (63,64).
Approaches targeting IGF-1R include two main groups: small-molecule IGF-1R TKIs, and the monoclonal antibodies (mAbs) directed against the extracellular domain. Most IGR-1R are in preclinical or an early clinical phase of development in advanced solid tumors. Most mAbs are in the early phase of clinical development in advanced NSCLC, but one of them, figitumumab (CP-751,871), is already being developed in phase III studies.
Figitumumab (CP-751,871)
Figitumumab is a fully human immunoglobulin (IgG2) mAb directed against the IGF-1R that was found to be over-expressed in some subsets of NSCLC. Figitumumab selectively binds to IGF-1R, preventing IGF1 from binding to the receptor and activating subsequent receptor autophosphorylation.
A phase I study was conducted to determine the recommended phase II dose of figitumumab in combination with paclitaxel and carboplatin in patients with advanced solid tumors (65). A total of 42 patients, including 35 with stages IIIB and IV NSCLC, were enrolled in eight dose-escalation cohorts. A MTD was not identified. Treatment was well tolerated. Fifteen objective responses were reported, including two CRs in NSCLC and ovarian cancer. Notably, levels of bioactive IGF-1 seemed to influence response to treatment, with objective responses in patients with a high baseline-free IGF-1 to IGFBP3 ratio seen only in the 10 and 20 mg/Kg dosing cohorts. Based on its favorable safety, pharmacokinetic and pharmacodynamic properties, the maximal feasible dose of 20 mg/Kg was selected for further investigation.
In a randomized phase II study, CT-naïve patients with advanced NSCLC were randomly assigned 2:1 to paclitaxel 200 mg/m2, carboplatin AUC6 and figitumumab at doses of 10 or 20 mg/Kg (PCF10, PCF20) or paclitaxel and carboplatin alone (PC) every 3 weeks for up to six cycles (66). Patients receiving the experimental treatment with response or SD were eligible to continue figitumumab as single agent until disease progression, while patients in the control arm who were considered to have PD were eligible to receive figitumumab as single agent or in combination with the same CT at the discretion of the investigator. A total of 156 patients were randomized, followed by the additional non-randomized single-arm cohort of another 30 patients with non-adenocarcinoma NSCLC enrolled to CT with the higher dose of figitumumab. The primary end-point was ORR. In the randomized portion of the study, an ORR of 54% was observed in the PCF arm, against 42% in the CT-alone arm (P<0.0001). Particularly impressive was the response rate of 78% among 9 patients with squamous NSCLC treated with CT and figitumumab at a dose of 20 mg/Kg (57% in patients with adenocarcinoma). An apparent dose-response relation was observed with an ORR of 57% and 38% for squamous cell and adenocarcinoma, respectively, with a dose of 10 mg/kg. Similar results were observed in the analysis of clinical benefit response: the numerically highest clinical benefit, 89%, was observed in patients with squamous cell tumors treated with PCF20. Clinical benefit was also observed in three patients with squamous histology receiving figitumumab after progression with CT alone. In the non-randomized single-arm extension cohort, 16 of 23 patients assessable for efficacy, including 11 of 14 patients with squamous histology (79%), also responded to treatment. Median PFS in the PCF10 and PCF20 cohorts were 3.6 and 5 months, respectively. Of note, the median PFS of patients treated with PC, regardless of whether they received figitumumab maintenance, was 4.3 months. This translated to a PCF20/PC HR of 0.8 that was not significant (P=0.07). However, it is important to note that 37% of patients treated with PC received figitumumab. When patients treated with PC receiving figitumumab were censored for progression, the PC and PCF10 PFS curves superimposed each other, and a PFS advantage with an HR of 0.56 (95% CI, 0.28 to 0.87; P=0.0153) was observed for PCF20 compared with PC alone. PCF was well tolerated, with hyperglycemia, fatigue and neutropenia being the more frequent side effects.
The reported results underlined the major effect of figitumumab in non-adenocarcinoma histology, which may be related to a differential expression or activity of IGF-1R across NSCLC subtypes. In this respect, multiple lines of evidence indicate that deregulation of the IGF-1R pathway may be common in NSCLC of squamous histology. Low serum levels of IGFBP3, which controls the bioactivity of IGF-1, were reported in patients with squamous cell carcinoma (67). Thus, the further development of this drug, in each line of therapy, was addressed specifically to this NSCLC subtype.
Based on the previous clinical findings, a phase III trial, ADVIGO 1016 (Advancing IGF-1R in Oncology) was performed A total of 820 patients with advanced non-adenocarcinoma were to be randomized 1:1 to first-line carboplatin plus paclitaxel and figitumumab 20 mg/Kg (PCF) or paclitaxel and carboplatin alone (PC). However, this trial stopped accrual at 681 patients enrolled, due to a planned interim analysis at 225 events, which showed that the addition of figitumumab to PC would be unlikely to meet the primary end-point of improving OS, compared with PC alone (68). An apparent imbalance of certain serious AEs between the treatment arms, with more events, including fatalities, occurring in patients who were randomized to receive figitumumab, was observed. Serious AEs in the PCF arm included dehydration, hyperglycemia and hemoptysis. The potential relationship with early death (within 42 days of randomization) of a series of clinical and laboratory parameters was investigated. Low pre-treatment body mass index (P=0.003) and creatinine clearance (P=0.1) were predictive of early death for patients receiving figitumumab. Furthermore, PCF/PC survival HR estimates favored PC in patients with low baseline IGF-1 and PCF in those with high baseline IGF-1.
ADVIGO 1017 is another phase III trial that will evaluate figitumumab in combination with a different CT regimen, cisplatin and gemcitabine, as first-line treatment of advanced NSCLC. This study is still in the planning stage and will incorporate lessons learned from ADVIGO 1016 into its final design.
Other anti-IGF-1R mAbs
Several other anti-IGF-1R mAbs in the treatment of advanced solid tumors are being investigated. Here, we report those for which the available results can be considered interesting enough to warrant further investigation in an early phase of clinical development of advanced NSCLC.
IMC-A12 (cixutumumab) is a fully human IgG1 mAb that binds with high affinity to the IGF-1R, thereby inhibiting ligand-dependent receptor activation and the subsequent activation of the PI3K/AKT signaling pathway. Although promising single-agent activity was observed in preclinical studies, the most impressive effects of targeting the IGF-1R with cixutumumab were seen when it was combined with cytotoxic drugs or other targeted therapies (69). Based on this evidence, several clinical trials are ongoing. The combination of carboplatin plus gemcitabine plus cetuximab with or without cixutumumab is being investigated as first-line treatment of any advanced histologic subtype NSCLC patients. The primary end-point is the ORR. Carboplatin plus paclitaxel plus cetuximab with or without cixutumumab is also being investigated in another phase II randomized trial as first-line metastatic therapy for any histologic subtype NSCLC. A third phase II trial is investigating the combination of carboplatin plus paclitaxel plus bevacizumab with or without IMC-A12 as first-line therapy for advanced non-squamous NSCLC patients. PFS is the primary objective for the last two trials. Cixutumumab is also being evaluated as second-line treatment in a phase II randomized trial comparing erlotinib with or without IMC-A12 in patients with any histologic subtype.
MK-0646 (dalotuzumab) is a humanized IgG1 mAb against IGF-1R, which is under development in advanced NSCLC. Two dose-finding trials evaluated the intravenous administration of MK-0646 either weekly or bi-weekly in advanced solid tumors (70,71). In a phase II randomized trial, called IMPACT, patients affected by advanced non-squamous NSCLC are randomized to receive cisplatin plus pemetrexed with or without weekly MK-0646 as first-line therapy. Another phase I/IIa trial is evaluating MK0646 in combination with erlotinib for patients with recurrent NSCLC.
A recombinant, human mAb directed against the IGF-1R is under investigation within a phase I dose-escalation trial aimed at CT-naïve patients. In this study, advanced NSCLC patients receive the combination of carboplatin plus paclitaxel plus BIIB022, all given intravenously, with the aim of determining their activity and safety in this setting.
|
|
Hepatocyte growth factor receptor inhibitors
The multifunctional growth factor scatter factor/hepatocyte growth factor (SF/HGF) and its receptor TK c-Met have emerged as a well-characterized ligand-receptor complex involved in multiple cell functions, including proliferation, invasiveness, cell migration, survival and angiogenesis (72). C-Met can be dysregulated through various mechanisms that include, but are not limited to, over-expression, gene amplification and mutation (73). New agents targeted against the c-Met kinase receptor or its ligand are now in the clinic and have shown promising results in several diseases. In particular, MET amplification has been documented in NSCLC, especially after treatment with EGFR TKIs. Indeed, about 20% of patients with an EGFR mutation who initially respond well to an oral EGFR inhibitor are found to have a c-Met mutation, and it is currently believed that this mutation contributes to “acquired resistance” to these agents when patients progress over time (73,74). Based on these considerations, dual EGFR-MET inhibition is a promising strategy for overcoming MET-mediated resistance to EGFR inhibitors. As a result, several c-Met inhibitors are under development in combination with EGFR inhibitors, such as erlotinib, in ongoing clinical trials.
ARQ 197-209 is an orally administered, selective, non-ATP competitive inhibitor of c-Met. A randomized phase II study with this agent was presented at the 2010 ASCO Annual Meeting (75). One hundred and sixty-seven patients with advanced NSCLC of any histology, previously treated with a single line of CT and EGFR inhibitor-naïve, were randomized to receive erlotinib at the typical 150 mg daily dose plus ARQ 197 360 mg twice daily or erlotinib plus placebo. Archival tissue was collected for all patients for k-Ras, EGFR and c-Met analyses. The primary end-point of the trial was PFS; secondary end-points included safety, ORR, OS and sub-group analyses. PFS was 66% longer in patients who received ARQ 197 with erlotinib (median 16.1 weeks) than in those who received second-line erlotinib alone (median 9.7 weeks). This was not a statistically significant difference (HR=0.81; 95% CI, 0.57-1.15; P=0.23), but a pre-specified analysis in patients with non-squamous histology (n=117) showed a statistically significant improvement in PFS for patients who received ARQ197 plus erlotinib over those treated with erlotinib plus placebo (median PFS, 18.9 vs 9.7 weeks; HR=0.61; P<0.05). PFS improvement was particularly prominent among patients with non-squamous histology, EGFR wild-type status and K-ras mutations. Safety analysis revealed no major differences between arms with AEs (>10% of patients; all grades), including rash, diarrhea, fatigue, nausea and anemia.
XL184 is a potent orally available inhibitor of c-Met, Ret, Kit and VEGFR2, which is being evaluated in a phase I/II trial. In phase I of the study, the purpose is to evaluate the safety, tolerability and highest safe dose of XL184 in combination with erlotinib administered to patients with NSCLC. In phase II, the purpose is to evaluate the ORR of daily oral administration of XL184 with or without erlotinib in patients with NSCLC and documented progressive disease, following a prior RECIST response to monotherapy with erlotinib or following SD of at least 6 months on monotherapy with erlotinib.
|
|
Inhibition of intracellular signaling pathways
Targeting the Ras/MAPK pathway
The Ras/MAPK pathway is involved in cell proliferation and inhibition of apoptosis. Inappropriate oncogenic activation of the MAPK pathway, such as by Ras, is a feature of many neoplasms, including NSCLC. Various components of this pathway can be interrupted for therapeutic purposes, and preliminary data are available for some of these strategies.
Ras
Oncogenic Ras mutations have been identified in approximately 30% of human cancers, with K-Ras mutations occurring in 40% of NSCLC (76). Ras can be inhibited by antisense molecules (eg ISIS 2503), farnesyl transferase inhibitors (FTIs) and peptide vaccines.
The enzyme farnesyl transferase is involved in postranslational modification of the Ras proteins by covalently linking a farnesyl group. This permits the Ras protein to be translocated to the surface membrane, allowing the protein to be involved in signaling for increased proliferation and inhibition of apoptosis (77). Single agent activity in NSCLC, both alone and in combination with standard CT, has been reported in phase I studies using various FTI, including tipifarnib and lonafarnib (78,79). The dose-limiting toxicities of these agents were fatigue, myelosuppression and neurotoxicity. To determine whether these FTIs have clinical activity in NSCLC, several phase II clinical trials have been performed (80,81). Disappointing clinical activity was noted in a phase II study of 300 mg tipifarnib administered orally twice daily for 21 of every 28 days in 44 patients with previously untreated advanced NSCLC (80). No objective CR or PR were documented. Seven patients (16%) had SD for longer than 6 months. Median PFS was 2.7 months and median OS was 7.7 months. However, results of a second phase II study demonstrated that the combination of oral lonafarnib at a continuous dose of 100 mg twice per day with 175 mg/m2 paclitaxel every 3 weeks had considerable effect on 33 patients with taxane-refractory/resistant metastatic NSCLC (81). PR was achieved in 3 patients (10%) and 11 patients (38%) had sustained SD for ≥ 4 cycles of treatment. The median OS time was 39 weeks and the median PFS time was 16 weeks. Treatment was safe and the majority of AEs were moderate in severity and manageable. These results suggest that the future development of TKIs in NSCLC should be in combination with cytotoxic agents. A phase II trial to study the effectiveness of combining tipifarnib with gemcitabine and cisplatin in treating patients with advanced NSCLC is ongoing.
Raf kinase
Raf kinase can be inhibited by antisense molecules (eg ISIS 5132) and the Raf-1 kinase sorafenib. In a phase II study, ISIS 5132 showed no significant evidence of clinical activity in patients with advanced NSCLC (82).
Targeting the PI3K/AKT/mTOR pathway
The deregulated PI3K/AKT/mTOR signaling pathway is reported to contribute to lung cancer development and maintenance. In particular, several preclinical data support the primary role of the PI3K pathway in proliferation, survival, disease progression and resistance to chemo and radiotherapy in NSCLC cell lines (83). Frequent Akt activation and mTOR phosphorylation were found in 51% of NSCLC patient samples and in 74% of NSCLC cell lines. Moreover, both PI3KCA amplification and, to a lesser extent, PI3KCA mutations are found in NSCLC (84,85). The phosphatase and tensin homologue gene (PTEN) is a tumor supressor gene, involved in the regulation of the PI3K pathway. There is evidence that PTEN dephosporylates 3-phosphatidylinositol 3,4,5-trisphophate (PIP3) while mutated PTEN cannot. Therefore, PTEN negativey regulates the PI3K/Akt/mTOR pathway, and cancer cells in which the PTEN gen is deleted or its expression is downregulated display constitutively activated PI3K signaling, which contributes to lung carcinogenesis (86). PTEN may be downregulated through several mechanism, including mutations, loss of heterozygosity, methylation, aberrant expression of regulatory microRNA, and protein instability. mTOR plays a critical role in transducing proliferative signaling mediated through the PI3K and Akt signaling pathways, principally activating downstream protein kinases that are required for both ribosome biosynthesis and translation of protein mRNAs that are essential for G1 to S phase transverse. Its inappropriate activation is involved in the pathogenesis of numerous tumor types, including NSCLC. Thus, it is a key target to block by pharmacological inhibition, as a strategy for the development of anticancer agents (87).
Numerous drugs interfere with the PI3K/AKT/mTOR pathway at multiple levels and may be active even in the absence of a PI3KCA mutation owing to frequent alterations at various levels of this pathway, such as PTEN loss, Akt activation, etc. PI3K inhibitors have shown efficacy in vitro and are currently being tested in early phase clinical trials (88). Compounds targeting the mTOR pathway include rapamycin and its derivatives CCI-779 (temsirolimus) and RAD001 (everolimus).
Rapamycin (Sirolimus)
Rapamycin, developed initially as an antifungal drug, also possesses immunosuppressive and antiproliferative properties. It was efficacious in inhibiting the growth of human NSCLC cells. In animal models, it effectively inhibited the growth of an NSCLC tumor and alveolar epithelial neoplasia induced by Ras (89). Evidence that the combination of rapamycin and docetaxel is synergistic in inhibiting the growth of lung cancer cells (90) led to the hypothesis that mTOR inhibitors could be more efficacious when combined with other therapies, such as CT or other targeted agents, in lung cancer treatment. No clinical data on rapamycin in the treatment of NSCLC are available, but it is being tested in combination with pemetrexed for previously treated patients.
CCI-779 (Temsirolimus)
Temsirolimus, a water-soluble ester of rapamycin, showed significant antitumor activity in preclinical and phase I studies in a variety of human cancer models, including NSCLC (91,92).
CCI-779 at two weekly iv doses (25 and 250 mg) was tested as maintenance treatment in a phase II trial of 86 patients affected by extensive-stage small-cell lung cancer in remission after CT induction (93). It appears to have significant activity, with a median PFS time of 2.2 months and a median OS of 7.8 months.
Another two-stage single arm phase II trial evaluated the response and toxicity rates of temsirolimus administered as front line single agent treatment for stage III (pleural effusion) or IV NSCLC (94). A total of 55 patients received 25 mg of temsirolimus administered intravenously as a 30 minute infusion on days 1, 8, 15 and 22 in 4-week cycles. Results were interesting, with four (8%) confirmed PRs and 15 (30%) patients with SD, making the total DCR 38%. The median PFS time was 2.3 months and the median OS time was 6.6 months. The most common toxicity was grade 3-4 dyspnea (12%), fatigue (10%), hyperglycemia (8%), hypoxia (8%), nausea (8%) and rash-desquamation (6%). Although the study did not meet the predefined success criteria, temsirolimus had good tolerability and similar activity to other signal transduction inhibitors.
RAD001 (Everolimus)
Everolimus is an orally available rapamycin analogue with antitumor activity. Everolimus as a single agent and in combination with other anticancer agents showed efficacy in cancer cell lines and xenograft NSCLC models, as well as in phase I studies (95).
Everolimus monotherapy at an oral dose of 10 mg/day until progression has also been evaluated in a phase II trial involving 85 patients with refractory advanced NSCLC (96). All patients were refractory to platinum-based treatment and were enrolled in two separate treatment arms: patients previously treated with two or fewer CT (arm 1; n=42) and patients previously treated with two or fewer CT and EGFR-TKIs (arm 2; n=43). The ORR was 4.7% (7.1% in arm 1 and 2.3% in arm 2), with an overall DCR of 47.1%. Median PFS was 2.6 months in arm 1 and 2.7 months in arm 2. Treatment was well tolerated, with the main grade 3 and 4 toxicities being fatigue and dyspnea in arm 1 and mucositis, hypokalemia and hyponatremia in arm 2. Overall, this approach was thought to have not enough single agent activity and was discontinued.
The pharmacodynamic effects of RAD001 in patients with recurrent NSCLC have been evaluated by FDG-PET (97). In eight patients receiving oral RAD001 at 10 mg daily, a FDG-PET scan was performed at baseline and after 8 days. A reduction in the sum of the maximum standardized uptake value (SUV max) on day 8 was observed in all patients, suggesting FDG-PET as a potential tool for early evaluation of the pharmacodynamic effect of RAD001 in patients with NSCLC.
Patient selection for treatment with mTOR inhibitors
Although mTOR signaling is commonly deregulated in cancer, mTOR inhibitors have failed to show any appreciable single activity in many tumor types. Based on preclinical data, a variety of predictors of response have been proposed, but most have not yet been clinically validated. Patients with decreased PTEN may specially benefit from rapamycin analogs. mTOR inhibition reduces neoplastic proliferation and tumor size in PTEN + mice, demonstrating that mTOR is the major effector of oncogenic PI3K signaling (98). However, the predictive role of PTEN in clinical trials remains controversial (99). Activation of PI3K signaling, regardless of mechanism (PTEN loss or activated receptor-tyrosine-kinase signaling), may sensitize tumors to mTOR inhibition (100). Tumor growth conferred by Akt activation is also reversed by mTOR inhibitors. Rapamycin analogs also block tumor growth induced by oncogenic PI3KCA mutations, suggesting that activating PI3K mutations may also have predictive value (101). Finally, stimulation of the insulin and IGF-1R activates the PI3K/Akt/mTOR pathway causing pleiotropic cellular effects including an mTOR-dependent loss in insulin receptor substrate-I expression leading to feedback down-regulation of signaling through the pathway (102). Feedback inhibition could have marked biological and therapeutics implications. First, feedback inhibition of upstream signaling pathways could cause hypersensitivity to mTOR inhibitors and inhibition of other elements of the activated signaling pathway (so-called “oncogene addiction”) (103). Second, inhibition of mTOR could cause the release of feedback inhibition, paradoxically activating IGF-I signaling and reducing the antitumor effects of mTOR inhibitors. It has been shown that inhibition of mTOR in cancer cell lines and in patient tumors causes activation of Akt kinase which is prevented by IGF-IR inhibition. Furthermore, IGF-I antagonizes the antiproliferative affects of rapamycin analogs and IGF-1R inhibitors sensitize cancer cell lines to rapamycin´s antiproliferative effects (102).
In conclusion, there remains an urgent need to better understand mTOR inhibitors´s mechanism of action and to identify predictive markers of response that can be used to prospectively select patients who will derive the greatest benefit from rapamycin analogs.
|
|
EML4-ALK fusion oncogene
The anaplastic lymphoma kinase (ALK) gene is frequently involved in translocations that lead to gene fusions in a variety of human malignancies, including lymphoma and lung cancer. Fusion partners of ALK include NPM, EML4, TPM3, ATIC, TFG, CARS and CLTC. Characterization of ALK fusion patterns was identified in 2007 in Japanese NSCLC and their resulting clinicopathological profiles could be of great benefit in better understanding the biology of lung cancer (104).
A group of patients with NSCLC have tumors that contain an inversion in chromosome 2 (Inv(2)(p21p23)) that joins exons 1-13 of EML4 to exons 20-29 of ALK. The resulting chimeric protein, EML4-ALK, contains an N-terminus derived from EML4 and a C-terminus containing the entire intracellular tyrosine kinase domain of ALK. This fusion oncogene rearrangement is transforming both in vitro and in vivo and defines a distinct clinico-pathological subset of NSCLC.
The oncogenic role of the EML-ALK4 fusion oncogene provides a potential avenue for therapeutic intervention. Cancer cell lines harboring the EML4-ALK translocation are effectively inhibited by small molecule inhibitors that target ALK (105).
ALK gene rearrangements or the resulting fusion proteins may be detected in tumor specimens using immunohistochemistry (IHC), reverse transcription polymerase chain reaction of cDNA (RT-PCR) and fluorescence in situ hybridization (FISH).
The clinico-pathological features of EML4-ALK-positive adenocarcinoma are reported to include its high incidence in young (median age of 53 years compared with 66 years in other lung cancer patients), non-smoking patients, tumors that show distinct solid or acinar growth patterns with or without signet-ring cell histology, and its mutually exclusive occurrence with mutations in EGFR and KRAS (106).
In unselected NSCLC populations, the EML4-ALK rearrangement is a relatively rare event, present in about 4% of lung adenocarcinomas and essentially limited to never smokers, equivalent to over 70,000 patients diagnosed annually worldwide.
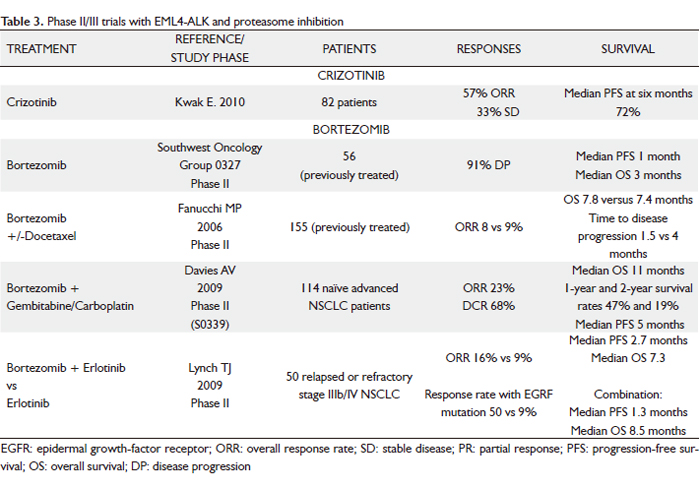
The initial studies reporting on the discovery of EML4-ALK raised the possibility that inhibiting the kinase activity of ALK may be an effective clinical therapy. Furthermore, transgenic mice expressing EML4-ALK in the lung epithelium develop numerous lung adenocarcinomas, demonstrating the oncogenic nature of this fusion gene. Pre-clinical studies demonstrate that EML4-ALK NSCLC cell lines undergo down-regulation of critical survival signaling pathways and apoptosis when treated with an ALK kinase inhibitor. This is analogous to what has been observed with EGFR inhibitors in EGFR mutant NSCLC. Similarly, ALK inhibitors have been evaluated in vivo in xenograft models generated from EML4-ALK NSCLC cell lines and lead to effective regressions of established tumors. Currently, only one agent targeting ALK, PF-02341066 initially designed as an inhibitor of MET, is in clinical use, although others have been examined in pre-clinical model systems (107).
The small molecule TKI crizotinib (PF02341066) is an orally bioavailable ALK inhibitor of phosphorylation and signal transduction. This inhibition is associated with G1-S phase cell cycle arrest and induction of apoptosis in positive cells in vitro and in vivo.
The phase I study of this agent started in May 2006. After screening tumor samples from approximately 1500 patients with NSCLC for the presence of ALK rearrangements, authors identified 82 patients with advanced ALK-positive disease who were eligible for the clinical trial. Most of the patients had received previous treatment. These patients were enrolled in an expanded cohort study instituted after phase 1 dose escalation had established a recommended crizotinib dose of 250 mg twice daily in 28-day cycles. Patients with ALK rearrangements tended to be younger than those without the rearrangements, and most of them had little or no exposure to tobacco and had adenocarcinomas. At a mean treatment duration of 6.4 months, the ORR was 57% (47 of 82 patients, with 46 confirmed PR and 1 confirmed CR); 27 patients (33%) had SD. A total of 63 of 82 patients (77%) were continuing to receive crizotinib at the time of data cutoff, and the estimated probability of 6-month PFS was 72%, with no median for the study reached. The most common side effects were fatigue, nausea/vomiting, diarrhea and visual disturbances associated with the transition from dark to light (108).
These dramatic findings led to two subsequent clinical trials of PF-02341066. The first is a randomized phase III trial of PF-02341066 compared with standard second line CT (pemetrexed or docetaxel) in EML4-ALK NSCLC and will be accruing at 179 sites worldwide. The second is a phase II clinical trial of single agent PF-02341066 in EML4-ALK NSCLC designed for patients not eligible for the phase III trial or patients randomized to CT who subsequently developed PD.
EML4-ALK NSCLC is a unique subset of NSCLC patients for whom ALK inhibitors may represent a very effective therapeutic strategy. The challenge remaining is to incorporate and disseminate widespread use of diagnostic testing for EML4-ALK to identify this patient subset; thus, it is essential to screen patients by genetic testing and not rely solely on the presence of clinical predictors. The results of genetic screening can then be used to choose the appropriate molecularly targeted therapy (Table 3).
 |
|
Bortezomib (proteasome inhibition)
Bortezomib (PS-341, Velcade, Millennium Pharmaceuticals, Inc.) is a dipeptidyl boronic acid that functions as a specific and selective reversible inhibitor of the 26S proteasome. The ubiquitin-proteasome pathway plays an important role in the regulation of cell proteins, with regard to cell cycle control, transcription, apoptosis, cell adhesion, angiogenesis and tumor growth.
Bortezomib is the first proteasome inhibitor evaluated in clinical trials. In vitro experiments have shown that this treatment has a cytotoxic effect on various breast, colorectal, ovarian, pancreatic, prostate, lung and oral cancer cells. Although bortezomib has shown its greatest benefit in the treatment of refractory multiple myeloma, it targets many key cell cycle regulators that are relevant to tumor progression and therapy resistance in lung cancer. As a single agent, it has limited activity, but in combination with CT showed encouraging activity without significantly adding to toxicity (109).
Bortezomib can facilitate apoptosis by decreasing levels of Bcl-2 and Bcl-xL. They are reported to be over-expressed in up to 90% of small cell lung cancer (SCLC) tumors and are associated with CT resistance. The ability of bortezomib to overcome Bcl-2–mediated resistance to apoptosis and to stabilize the proapoptotic Bax, a binding partner of Bcl-2, are two of the proposed mechanisms by which bortezomib is thought to be of potential therapeutic benefit, particularly in SCLC. Preclinical data suggest that proteasome inhibition may reverse platinum resistance, which is a common cause of treatment failure and disease progression in lung cancer.
Initial phase I studies showed modest single-agent activity with bortezomib in NSCLC, which formed the basis for subsequent research into this tumor type. Patients receiving bortezomib alone had an 8% RR and a 21% SD rate, which is comparable with other second-line therapies in advanced NSCLC (110).
Activity was also seen in the histological subtype of bronchiolo-alveolar cancer in early phase studies with bortezomib. Two ongoing trials are examining the use of bortezomib in bronchiolo-alveolar cancer with two schedules: an industry-sponsored trial with the standard twice-weekly schedule (1.3 mg/m2) and a California Cancer Consortium study using a weekly schedule (dosed at 1.6 mg/m2).
Despite a strong preclinical rationale for single-agent bortezomib in SCLC, efficacy was limited in a phase II trial for extensive-stage disease (Southwest Oncology Group 0327). In previously treated patients with platinum-sensitive and -refractory extensive stage SCLC, treatment with 1.3 mg/m2 bortezomib was given on days 1, 4, 8 and 11 of a 21-day cycle to determine RR, toxicity and survival. Patients with histologically confirmed SCLC, measurable disease, performance status 0-1 and previous treatment with platinum-based therapy were enrolled. They were stratified by platinum-sensitivity status: sensitive (relapse >90 days after platinum) or refractory (progression during either 111).
One platinum-refractory patient had a confirmed PR. Although this could be viewed as a positive finding, given that platinum-refractory patients rarely respond to treatment, the lack of a predictive biomarker in this setting limits its potential clinical utility, if only a small proportion of patients are likely to benefit from bortezomib. As shown in preclinical models, testing of PS-341 in combination with an apoptotic trigger such as CT is a rational clinical approach.
Another phase II trial of bortezomib in CT-naïve patients with advanced-stage NSCLC was terminated at the first stage after 14 patients enrolled at 4 institutions. No objective response was observed. Three patients (21%) had SD and received 8, 6 and 4 cycles of treatment; the duration of SD was 11.5, 4.2 and 3.4 months, respectively. Median PFS was 1.3 months (95% CI, 0.6-3.0 months); median OS was 9.9 months (95% CI, 2.2-27.0 months). Thus, though well tolerated, bortezomib monotherapy is not active in cohorts of CT-naïve, metastatic NSCLC.
Given the effect of proteasome inhibition on cell cycle regulation, some authors have postulated that the sequence in which bortezomib is given with other chemotherapeutics may affect efficacy. In cell types where bortezomib is principally cytostatic, pre-treatment may diminish chemotherapeutic activity, whereas concurrent treatment or post-treatment reducing levels of anti-apoptotic Bcl-2 and increasing levels of the tumor suppressor p27 may reinforce CT. To investigate this, the California Cancer Consortium conducted a multi-institutional randomized phase II trial of concurrent vs sequential docetaxel and bortezomib treatment. Arm 1 received bortezomib (1.6 mg/m2 i.v.) on days 1 and 8 and arm 2 received bortezomib day 2, both with docetaxel day 1. Prophylactic GCSF support was recommended. The primary end-point was RR, while PFS and OS were secondary end-points. Docetaxel plus bortezomib concurrently given on day 1 had a similar RR to sequential therapy, but resulted in OS exceeding prior published OS estimates for either the agent alone or in combination (112).
Then, a randomized phase II study of bortezomib alone (1.5 mg/m2 i.v. on days 1, 4, 8 and 11 of a 21-day schedule) or in combination with docetaxel (75 mg/m2 i.v. on day 1) in 155 previously treated patients with advanced NSCLC showed similar RR (8% for bortezomib alone vs 9% for the combination) and median survival (7.8 vs 7.4 months), with time to disease progression improved in patients receiving the combination (1.5 vs 4 months). Hematological toxicity was 4% vs 65% grade 3/4 neutropenia and 5% vs 13% grade 3 anemia for bortezomib vs bortezomib + docetaxel, respectively (113).
Another randomized phase II trial is ongoing in Europe of bortezomib vs pemetrexed vs the combination. Previously, a phase I study of two different schedules of bortezomib and pemetrexed in advanced solid tumors with emphasis on NSCLC was examined. Two separate dose-escalating arms (arm A and arm B) were conducted simultaneously. Patients received pemetrexed on day 1 (D1) (500-600 mg/m2 IV) every 21 days. In arm A, bortezomib was given twice weekly (0.7-1.3 mg/m2 on D1, 4, 8 and 11). In arm B, bortezomib was given weekly (1.0-1.6 mg/m2 on D1 and 8). Of the 16 patients with NSCLC, 2 (12.5%) had PR and 9 had SD, for a DCR of 68.8%. Recommended phase II doses for arm A were 500 mg/m2 pemetrexed and 1.3 mg/m2 bortezomib twice weekly. For arm B, the recommended doses were 500 mg/m2 pemetrexed and 1.6 mg/m2 bortezomib weekly (114).
The other phase II trial (S0339) was conducted in CT-naïve advanced NSCLC patients with first-line treatment with bortezomib plus gemcitabine/carboplatin. Patients with selected stage IIIB/IV NSCLC, performance status 0-1 and no history of brain metastasis received up to six 21-day cycles of 1000 mg/m2 gemcitabine, days 1 and 8, carboplatin area under curve 5.0, day 1, and 1.0 mg/m2 bortezomib, days 1, 4, 8 and 11. One-hundred-and-fourteen patients (52% adenocarcinoma, 85% stage IV) received a median of 3.6 treatment cycles. Median OS was 11 months; 1-year and 2-year survival rates were 47% and 19%, respectively. Median PFS was 5 months. RR was 23%, and DCR (responses + SD) was 68% (114). The most common grade 3/4 toxicities were thrombocytopenia (63%) and neutropenia (52%) (115).
However, the potential benefit of EGFR TKIs with bortezomib is of particular interest. A preclinical model showed activity for the combination of erlotinib and bortezomib in erlotinib-sensitive bronchiolo-alveolar cells. The efficacy of this combination was tested in advanced NSCLC in a randomized phase II study of erlotinib vs erlotinib plus bortezomib. RR was 16% for erlotinib vs 9% for the combination; DCR was 52 vs 45%, respectively. Insufficient activity was seen with erlotinib plus bortezomib in patients with relapsed/refractory advanced NSCLC to warrant a phase 3 study of the combination (116).
Although bortezomib alone has shown some antitumor activity in lung cancer, it is likely to have its greatest clinical benefit when given in combination with other therapeutics, for example with carboplatin/gemcitabine. However, a phase III trial is required to confirm expected results (Table 3).
|
|
Histone deacetylase inhibitors
Chromatin is composed of regular repeating units of nucleosomes in which deoxyribonucleic acid (DNA) has been conserved. The main components of chromatin are DNA, ribonucleic acid (RNA), which are negatively charged, associated proteins, including histones, positively charged, and non-histone chromosomal proteins, which are acidic at neutral pH. Acetylation of histones is one of the many post-translational modifications that occur in these DNA-packaging proteins, which generally leads to increased accessibility to promoter regions and increased transcription of genes in localized areas of chromosomes. This process is opposed by the histone deacetylase classes of enzymes (HDAC), which promote condensation of chromatin and repression of gene expression (117).
HDAC inhibitors are a new class of targeted anticancer agents, which block deacetylation function, causing cell-cycle arrest, differentiation and/or tumor apoptosis. HDAC inhibitors have powerful antitumor activity in human xenograft models, suggesting their usefulness as novel anticancer agents. Many studies have shown that HDAC inhibitors are relatively non-toxic to normal cells or tissues exhibiting selective cytotoxicity against a wide range of cancer cells. Several HDAC inhibitors, based on promising preclinical data, are currently being investigated in early phase clinical trials, both as single agents and in combination with other cytotoxic therapies, showing activity against several hematological and solid tumors.
HDAC inhibitors can be divided into six groups based on their structure. Specifically, some of these HDAC inhibitors enhance the cytotoxic effects of radiation by attenuating DNA repair and inducing apoptosis in human NSCLC cells and have a marked synergism of action with standard chemotherapeutic agents.
Multiple HDAC inhibitors are in clinical development to target a wide variety of malignancies, including entinostat (SNDX-275/MS-275), vorinostat (MK-0683/SAHA), N-Acetyldinaline (CI-994), pivanex (AN-9), romidepsin (FK-228/depsipeptide) and panobinostat (LBH589) (118).
Pivanex was administered at the dose of 2.34 g/m2/day for three consecutive days and repeated every 3 weeks to 47 refractory NSCLC patients in a phase II trial. The most common toxicities were transient grade 1–2 fatigue and nausea. Results were: 6.4% PR, 30% SD and median OS of 6.2 months with 1-year survival of 26%. In a phase I trial 12 pretreated NSCLC patients received pivanex at a dose of 2.5 g/m2 on days 1-3 in combination with docetaxel at a dose of 75 mg/m2 on day 4, every 3 weeks. PR was reported in three patients (25%). The study demonstrated that pivanex can be administered safely in combination with docetaxel. Now there is an ongoing phase IIb trial in which 225 patients with relapsed NSCLC will be randomized to pivanex plus docetaxel vs docetaxel (119).
In a phase II trial, N-Acetyldinaline (CI-994) was administered at continuous oral daily dose of 8 mg/m2 to 32 pre-treated NSCLC patients. Two patients (7%) achieved PR; and eight (28%), SD lasting more than 8 weeks. Median OS was 30 weeks. CI-994 treatment was well-tolerated, with grade 3–4 thrombocytopenia reported in five (15.6%) patients. In a phase II trial of CI-994 plus gemcitabine vs gemcitabine plus placebo a total of 180 patients were enrolled, with a RR of 3.5% and 3.8% for CI-994 and placebo arm, respectively. MS was 189 and 186 days, respectively. Another randomized phase II trial investigated carboplatin plus paclitaxel with CI-994 (4 mg/m2/day orally) or placebo in first-line advanced NSCLC patients, but results have not yet been published (120).
Of all these, the development of vorinostat for lung cancer is most advanced. Vorinostat is FDA-approved as oral monotherapy for the third-line treatment of cutaneous T-cell lymphoma with response rate of 30%. The most common side effects were diarrhea, fatigue, nausea, dysguesia and thrombocytopenia, but these were generally mild and were all reversible on cessation of therapy for 4–7 days.
In a phase II study, vorinostat was administered, at the continuously oral dose of 400 mg daily, to 14 pre-treated NSCLC patients. Seven patients experienced SD with a median time-to-progression (TTP) of 2.8 months and an MS of 6.5 months. Main toxicities were grades 3–5 vascular events, grade 4 neutropenia, grade 3 lymphopenia, fatigue and high alkaline phosphatases (121).
Based on the preclinical studies previously reported, a phase I study investigated, in order to assess the safety profile, the combination of vorinostat and carboplatin plus paclitaxel in patients with advanced solid malignancies. Vorinostat was administered orally once daily (400 mg) for 2 weeks or twice daily (300 mg) for 1 week, every 3 weeks. Paclitaxel (200 mg/m2) plus carboplatin (AUC 6) was administered on day 1 of each 21-day treatment cycle. Of 25 evaluable patients, 10 out of 19 patients with advanced-stage NSCLC had a PR. Both schedules of vorinostat were well tolerated (122).
Then, a phase II randomized, double-blinded placebo-controlled study evaluated the efficacy of vorinostat in combination with carboplatin and paclitaxel in patients with advanced-stage NSCLC. Patients with previously untreated stage IIIB or IV NSCLC were randomly assigned (2:1) to carboplatin (AUC 6) and paclitaxel (200 mg/m2 day 3) with either vorinostat (400 mg by mouth daily) or placebo. The primary end-point was comparison of the RR. The median number of cycles was four for both treatment arms. The confirmed RR was 34% with vorinostat vs 12.5% with placebo (P=0.02). There was a trend toward improvement in median PFS (6.0 months vs 4.1 months; P = 0.48) and OS (13.0 months vs 9.7 months; P = 0.17) in the vorinostat arm. Grade 4 platelet toxicity was more common with vorinostat (18% vs 3%; P<0.05). Nausea, emesis, fatigue, dehydration and hyponatremia were also more common with vorinostat (123).
However, the subsequent phase III randomized trial of 253 patients was terminated prematurely due to failure to demonstrate an improvement in response rate, PFS or OS in the vorinostat arm. Vorinostat enhances the efficacy of carboplatin and paclitaxel in patients with advanced NSCLC, but further trials are needed (124,125).
Other clinical trials are ongoing in small-cell lung cancer and in NSCLC with vorinostat in combination with other targeted agents. One particularly interesting combination is vorinostat plus erlotinib. The rationale for combining HDAC inhibitors with EGFR-TKIs is based on preclinical data that HDAC inhibition restores sensitivity to EGFR-TKIs after the development of resistance. Entinostat and romidepsin are similarly being tested in combination with erlotinib, and results of these trials may yield different results, as all of these agents have different activity against individual HDAC proteins.
HDAC inhibition is a promising therapeutic strategy for treatment of NSCLC. Although recent large trials of the HDAC inhibitor vorinostat failed to demonstrate a benefit in unselected patients with NSCLC, other ongoing trials with these and newer agents may help to identify a particular subgroup of patients for whom this therapy is the most appropriate (126) (Table 4).
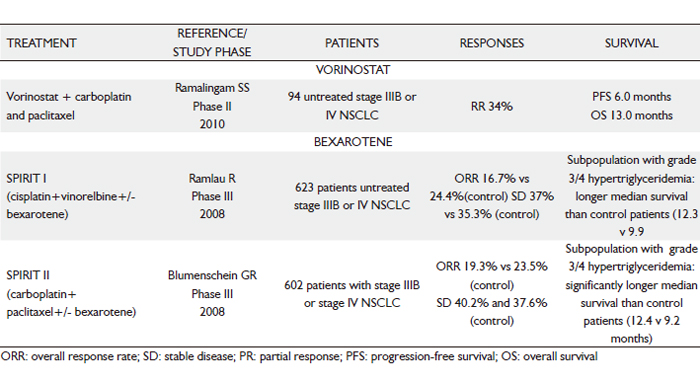 |
|
Retinoids
Several in vitro and in vivo studies have examined the positive and negative effects of retinoids (vitamin A analogs) in pre-malignant and malignant lesions. Retinoids have been used as chemopreventive and anticancer agents because of their pleiotropic regulator function in cell differentiation, growth, proliferation and apoptosis through interaction with two types of nuclear receptors: retinoic acid receptors (RAR) and retinoid X receptors (RXR). Recent investigations have gradually clarified the function of retinoids and their signaling pathways and may explain the failure of earlier chemopreventive studies (127).
There is a large body of literature on clinical and preclinical studies using natural retinoids and related compounds for the prevention and treatment of cancer. The field of lung cancer chemoprevention has been controversial until now. However, there has also been disappointment in extending the therapeutic use of bexarotene (selective RXR agonist) to patients with NSCLC. Bexarotene, a novel synthetic retinoid analogue, selectively binds to and activates RXRα, RXRβ and RXRγ; acts as a transcription factor to regulate the expression of genes responsible for cell proliferation and differentiation; and can lead to growth inhibition in hematopoietic and squamous cell tumor cell lines (128,129).
The phase I trials of bexarotene in patients with advanced cancer failed to produce objective responses in patients with NSCLC; they did, however, show some evidence of delayed TTP in these patients. Another phase I/II study of bexarotene in combination with cisplatin and vinorelbine in previously untreated advanced NSCLC reported the MTD of bexarotene as 400 mg/m2. It was to be administered continuously, beginning on day 1 and until progression of disease. All patients received 10 mg atorvastatin orally, beginning before bexarotene. Median survival was 14 months, and the projected 1- and 3-year survival rates were 61% and 30%, respectively. Hyperlipidemia was the most common toxicity associated with bexarotene therapy. These phase II results led to further investigation of bexarotene in combination with active CT regimens in a larger patient population in formal phase III trials. These trials, STI571 Prospective International Randomised Trial (SPIRIT) I and II, tested bexarotene in combination with carboplatin plus paclitaxel (CT) in CT-naïve patients with stage IIIB/IV NSCLC. SPIRIT I tested cisplatin plus vinorelbine with or without bexarotene, whereas SPIRIT II tested carboplatin plus paclitaxel with or without bexarotene. Totals of 623 patients (median age, 61 years) and 612 patients (median age, 63 years) were randomized to receive therapy in SPIRIT I and SPIRIT II, respectively. More than 80% of patients had stage IV disease. Adenocarcinoma was the most prevalent histological subtype (SPIRIT I, 42%; SPIRIT II, 50%-55%), followed by squamous cell carcinoma (SPIRIT I, 36%- 38%; SPIRIT II, 20%). Former and current smokers comprised 80% and 90% of patients in SPIRIT I and SPIRIT II, respectively.
In the SPIRIT I trial, patients treated with bexarotene plus CT had an OS of 8.7 months compared with 9.9 months in the control arm (P=0.3). Also, the respective 2-year survival rates (13.2% vs 15.7%; P=0.4) and PFS times (4.3 months vs 5 months; P=0.095) were worse with bexarotene. In the SPIRIT II trial, the median OS (9.2 months vs 8.5 months; P=0.2), the 2-year survival rate (16.3% vs 12.4%; P=0.2) and PFS (4.9 months vs 4.1 months; P=0.061) with CT alone vs bexarotene plus CT did not show any benefits of bexarotene therapy.
Based on the rationale that triglycerides are potential markers of the antitumor activity of bexarotene and rises in their levels may correlate with improved outcome, retrospective analyses were performed in subsets of patients reporting hypertriglyceridemia (National Cancer Institute grades 0-4). Pooled analysis from the SPIRIT I and II trials showed that 215 patients with grade 3/4 hypertriglyceridemia had a statistically longer survival than the control arm (hazard ratio = 1.31; P=0.0025). The independent multivariate survival analysis showed significantly longer survival in the bexarotene subset with hypertriglyceridemia (grade 0-4) than in control (SPIRIT I, P=0.0004; SPIRIT II, P=0.0002) (130).
One possible reason for these results is that solid tumors can acquire and develop intrinsic resistance to retinoids during carcinogenesis. The RXR selective compounds did show growth inhibitory effects when combined with the RAR retinoids. These results indicated that human lung cancer cell lines have a high degree of resistance to synthetic retinoids. The potential mechanisms of Retinoic Acid Resistance, i.e. increased P450 catabolism, drug export (P glycoprotein mediated), sequestration of retinoids by CRABs or other proteins, decreased expression of RARs through promoter methylation, persistent histone deacethylation, RAR rearrangement or mutation in the RAR ligand-binding domain and coactivator alteration or alterations downstream of target gene expression, may lead to cellular retinoid resistance. This knowledge should help predict patients most likely to benefit from retinoid therapy and develop strategies to optimize single agent or combination retinoid regimens to overcome resistance. The generation of retinoids and rexinoids with restricted selectivity has opened new possibilities for cancer therapy and chemoprevention. It is probable that demethylating and chromatin remodeling agents currently under clinical investigation could be combined with these new retinoids for a better restoration of RR expression (131).
Recently, a randomized phase II trial evaluated the combination of cisplatin and paclitaxel (PC) plus all-trans retinoic acid (ATRA) in patients with advanced NSCLC and its association with the expression of retinoic acid receptor beta 2 (RAR-beta2) as a response biomarker. Patients were assigned to receive ATRA 20 mg/m2/day or placebo, one week before treatment, until two cycles were completed. RAR-beta2 expression was analyzed in tumor and adjacent lung tissue. One hundred and seven patients were included, 55 in the placebo group and 52 in the ATRA group. RR for ATRA was 55.8% (95% CI, 46.6% to 64.9%) and for placebo, 25.4% (95% CI, 21.3 to 29.5%; P=0.001). The ATRA group had a longer median PFS (8.9 vs 6.0 months; P=0.008). Multivariate analysis of PFS showed significant differences for the ATRA group (hazard ratio, 0.62; 95% CI, 0.4 to 0.95). No significant differences in toxicity grade 3/4 were found between groups, except for hypertriglyceridemia (10% vs 0%) in ATRA. IHC and RT-PCR assays showed expression of RAR-beta2 in normal tissues of all tumor samples, but only 10% of samples in the tumor tissue. Thus, adding ATRA to CT could increase RR and PFS in patients with advanced NSCLC with an acceptable toxicity profile. A phase III clinical trial to confirm these findings is justified (132-134) (Table 4).
|
|
Conclusion
Lung cancer is the leading cause of cancer-related mortality in both men and women, with 1.2 million new cases diagnosed every year. Since most patients have advanced disease at diagnosis, CT is the mainstay of management, which has apparently reached a plateau of effectiveness in improving survival of NSCLC patients. Treatment outcomes in advanced or metastatic NSCLC remain unsatisfactory, with low long-term survival rates.
The major advances in the understanding the cancer biology and mechanisms of oncogenesis have allowed the development of several potential molecular targets for NSCLC treatments which are components of signalling pathways or metabolic processes which are relevant for cancer development and/or progression. A large amount of preclinical in vivo and in vitro data have been gathered on the antitumor properties of a number of new biological agents. Targeted inhibition of the VEGF or EGF signaling pathways has been clinically validated in the treatment of advanced NSCLC. Other anti-angiogenic drugs, such as sorafenib, sunitinib and a new class named 'vascular disrupting agents', are being tested in ongoing clinical trials, which will further define their role in the management of NSCLC. The ALK inhibitor crizotinib will become a key addition to the treatment of patients with NSCLC harboring genetic ALK translocations. The IGF-1R monoclonal antibody figitumumab, c-Met inhibitors and the HDAC inhibitor vorinostat or other agents in this class may have greater selectivity and efficacy.
It is important to note that in most of agents discussed, studies have been conducted similarly to that of clinical development of the classic chemotherapeutic drugs. It should be clear that the further development of molecular targets hinges on innovative studies with both serum and tumor tissue. But we don have to wait for negative results in phase III studies to search for subgroups. Predictive clinical characteristics and molecular biomarkers need to be identified early in developing in order to design phase II studies with selected population based on preclinical work. This would allow us identify agents that could be clinically useful without wasting time and resources with phase III trials based on modest phase II data in unselected population, as well as properly select patients who are most likely to benefit from treatment and to avoid unnecesary side effects to patients who would probably not receive any benefit. In conclusion, revising current classifications schemes to incorporate molecular features will better address the requirements of a targeted therapy approach within the context of personalized medicine, and enable researchers to use novel pathway inhibitors as an integral part of the therapeutic arsenal in the battle against lung cancer.
|
|
References
Cite this article as: Méndez M, Custodio A, Provencio M. New molecular targeted therapies for advanced non-small-cell lung cancer. J Thorac Dis 2011;3(1):30-56. doi: 10.3978/j.issn.2072-1439.2010.12.03
|

